
ESM Protein Function
Collection
Protein language models fine-tuned for function prediction using the Gene Ontology. • 12 items • Updated • 1
An Evolutionary-scale Model (ESM) for protein function prediction from amino acid sequences using the Gene Ontology (GO). Based on the ESM Cambrian Transformer architecture, pre-trained on UniRef, MGnify, and the Joint Genome Institute's database and fine-tuned on the AmiGO Boost protein function dataset, this protein language model predicts the GO subgraph for a particular protein sequence - giving you insight into the molecular function, biological process, and location of the activity inside the cell.
Sequence-to-function prediction — Predicts Molecular Function, Biological Process, and Cellular Component ontologies directly from raw amino acid sequences, eliminating the need for homology searches, structural data, or multiple sequence alignments.
Hierarchy-aware GO subgraph reconstruction — Outputs a full GO directed acyclic graph (DAG) ensuring predictions respect the ontology structure rather than treating each term as an independent binary label.
Efficient inference at scale — Supports weight quantization and quantization-aware training (QAT), enabling memory-efficient, high-throughput screening of large sequence datasets without accuracy loss.
"The Gene Ontology (GO) is a concept hierarchy that describes the biological function of genes and gene products at different levels of abstraction (Ashburner et al., 2000). It is a good model to describe the multi-faceted nature of protein function."
"GO is a directed acyclic graph. The nodes in this graph are functional descriptors (terms or classes) connected by relational ties between them (is_a, part_of, etc.). For example, terms 'protein binding activity' and 'binding activity' are related by an is_a relationship; however, the edge in the graph is often reversed to point from binding towards protein binding. This graph contains three subgraphs (subontologies): Molecular Function (MF), Biological Process (BP), and Cellular Component (CC), defined by their root nodes. Biologically, each subgraph represent a different aspect of the protein's function: what it does on a molecular level (MF), which biological processes it participates in (BP) and where in the cell it is located (CC)."
From CAFA 5 Protein Function Prediction
The code repository can be found at https://github.com/andrewdalpino/ESMC-Protein-Function.
The following pretrained models are available on HuggingFace Hub and require the esmc-protein-function library version 1.x.x for inference. All V1 models have been optimized with quantization-aware post-training.
| Name | Embedding Dimensions | Encoder Layers | Context Length | Total Parameters |
|---|---|---|---|---|
| andrewdalpino/ESMC-Protein-Function-V1-300M | 960 | 30 | 2048 | 397M |
| andrewdalpino/ESMC-Protein-Function-V1-600M | 1152 | 36 | 2048 | 661M |
The following pretrained models are available on HuggingFace Hub and require the esmc_function_classifier library version 0.1.x for inference.
| Name | Embedding Dimensions | Encoder Layers | Context Length | Total Parameters |
|---|---|---|---|---|
| andrewdalpino/ESMC-Protein-Function-V0-300M | 960 | 30 | 2048 | 361M |
| andrewdalpino/ESMC-Protein-Function-V0-300M-QAT | 960 | 30 | 2048 | 361M |
| andrewdalpino/ESMC-Protein-Function-V0-600M | 1152 | 36 | 2048 | 644M |
| andrewdalpino/ESMC-Protein-Function-V0-600M-QAT | 1152 | 36 | 2048 | 644M |
First, install the esmc-protein-function package using pip. I recommend using a virtual environment such as venv to keep dependencies compartmentalized.
pip install esmc-protein-function
Then, we'll load the model weights from HuggingFace Hub by calling the from_pretrained() method. We'll also need the ESM tokenizer from the esm library. Then, tokenize the sequence and query the model like in the example below.
import torch
from esm.tokenization import EsmSequenceTokenizer
from esmc_protein_function.model import ESMCProteinFunction
model_name = "andrewdalpino/ESMC-Protein-Function-V1-300M"
sequence = "MPPKGHKKTADGDFRPVNSAGNTIQAKQKYSIDDLLYPKSTIKNLAKETLPDDAIISKDALTAIQRAATLFVSYMASHGNASAEAGGRKKIT"
top_p = 0.5
tokenizer = EsmSequenceTokenizer()
model = ESMCProteinFunction.from_pretrained(model_name)
out = tokenizer(sequence, max_length=2048, truncation=True)
input_ids = torch.tensor(out["input_ids"], dtype=torch.int32)
go_term_probabilities = model.predict_terms(
input_ids, top_p=top_p
)
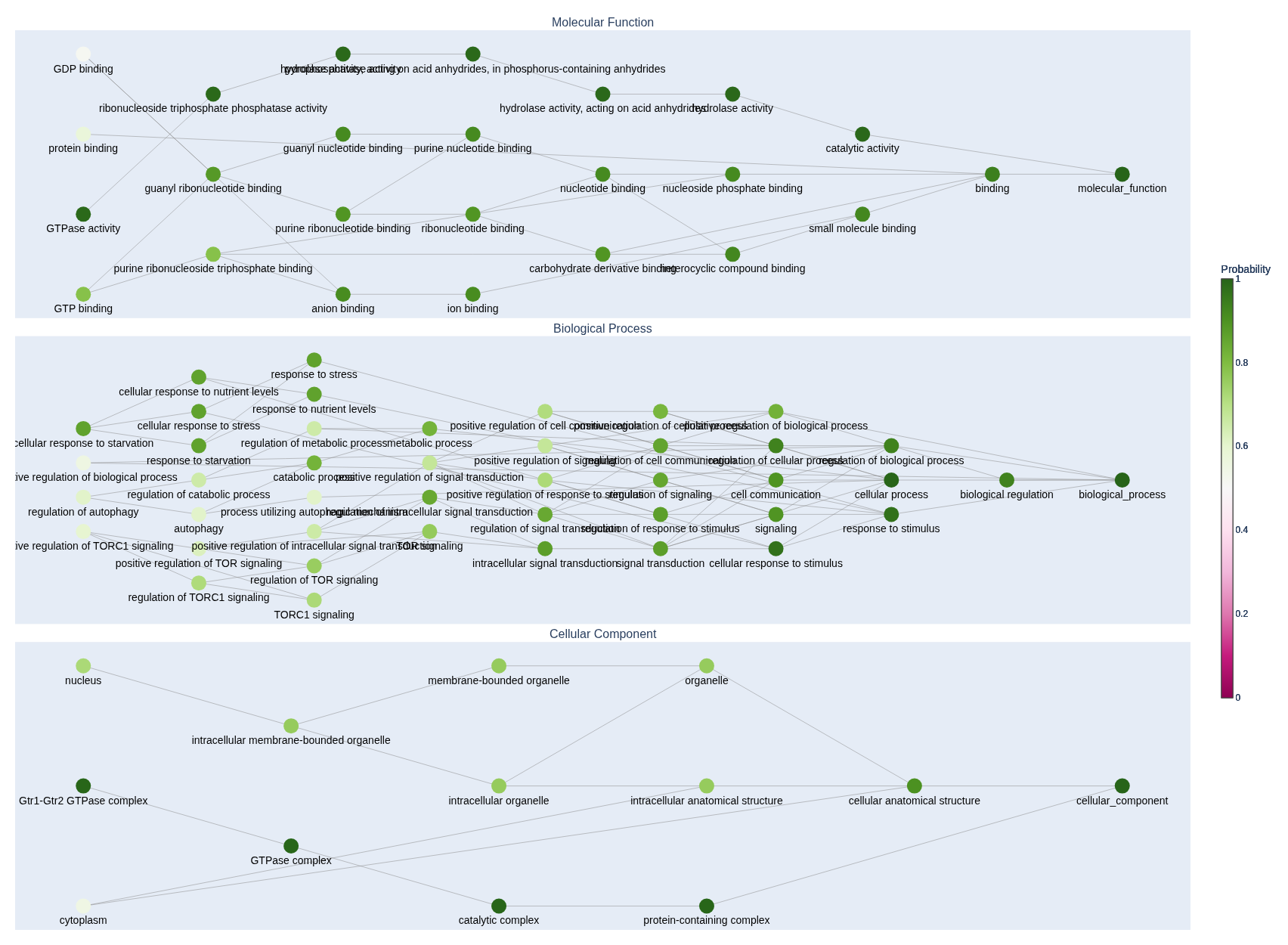
You can also output the gene-ontology (GO) networkx subgraph for a given sequence like in the example below. You'll need an up-to-date gene ontology database that you can import using the obonet package.
pip install obonet
Then, load the GO DAG and call the predict_all_subgraphs() method like in the example below.
import networkx as nx
import obonet
# Visit https://geneontology.org/docs/download-ontology/ to download.
go_db_path = "./dataset/go-basic.obo"
graph = obonet.read_obo(go_db_path)
model.load_gene_ontology(graph)
subgraphs, go_term_probabilities = model.predict_all_subgraphs(
input_ids, top_p=top_p
)
# Render the subgraphs ...
To quantize the model weights using int8 call the quantize_weights() method. Any model can be quantized, but we recommend one that has been quantization-aware trained (QAT) for the best performance. The group_size argument controls the granularity at which quantization scales are computed. Choose a group size that can divide the embedding dimensions equally.
model.quantize_weights(group_size=64)
- T. Hayes, et al. Simulating 500 million years of evolution with a language model, 2024.
- M. Ashburner, et al. Gene Ontology: tool for the unification of biology, 2000.
- Z. Lin, et al. A Structured Self-attentive Sentence Embedding, ICLR 2017.