Spaces:
Running
on
T4

Running
on
T4
File size: 8,794 Bytes
e1a6cd9 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 |
# ProteinMPNN
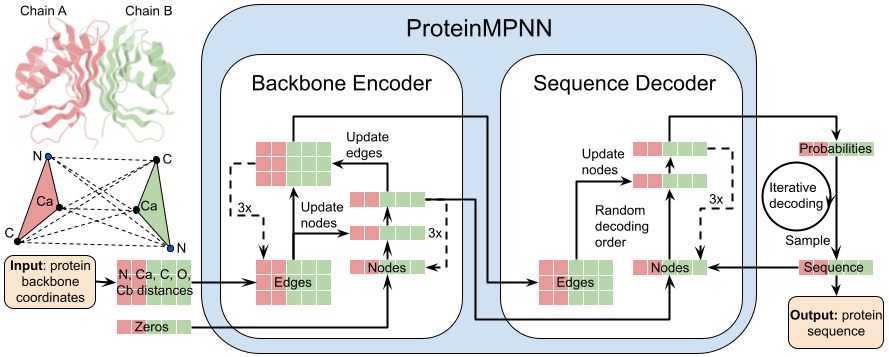
Read [ProteinMPNN paper](https://www.biorxiv.org/content/10.1101/2022.06.03.494563v1).
To run ProteinMPNN clone this github repo and install Python>=3.0, PyTorch, Numpy.
Full protein backbone models: `vanilla_proteinmpnn`.
Code organization:
* `vanilla_proteinmpnn/protein_mpnn_run.py` - the main script to initialialize and run the model.
* `vanilla_proteinmpnn/protein_mpnn_utils.py` - utility functions for the main script.
* `vanilla_proteinmpnn/helper_scripts/` - helper functions to parse PDBs, assign which chains to design, which residues to fix, adding AA bias, tying residues etc.
* `vanilla_proteinmpnn/examples/` - simple code examples.
-----------------------------------------------------------------------------------------------------
Input flags:
```
argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;")
argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise")
argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-mean[log_probs] to npy files")
argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position")
argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs")
argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)")
argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)")
argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms during the inference.")
argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target.")
argparser.add_argument("--batch_size", type=int, default=1, help="Batch size when using GPUs.")
argparser.add_argument("--max_length", type=int, default=20000, help="Maximum sequence length.")
argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.1 0.3 0.5. Sampling temperature for amino acids, T=0.0 means taking argmax, T>>1.0 means sampling randomly.")
argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/")
argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed.")
argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB.")
argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed PDBs into jsonl.")
argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.")
argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions.")
argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.")
argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.")
argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.")
argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices.")
argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm.")
argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions.")
argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs.")
argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True.")
argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True.")
argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions for symmetric design.")
```
-----------------------------------------------------------------------------------------------------
Example from `vanilla_proteinmpnn/examples/` to design a single PDB file:
```
path_to_PDB="../PDB_complexes/pdbs/3HTN.pdb"
output_dir="../PDB_complexes/example_3_outputs"
if [ ! -d $output_dir ]
then
mkdir -p $output_dir
fi
chains_to_design="A B" #design only chains A and B while using the context of other chains
python ../protein_mpnn_run.py \
--pdb_path $path_to_PDB \
--pdb_path_chains "$chains_to_design" \
--out_folder $output_dir \
--num_seq_per_target 2 \
--sampling_temp "0.1" \
--batch_size 1
```
-----------------------------------------------------------------------------------------------------
Example from `vanilla_proteinmpnn/examples/` to design some monomers:
```
folder_with_pdbs="../PDB_monomers/pdbs/"
output_dir="../PDB_monomers/example_1_outputs"
if [ ! -d $output_dir ]
then
mkdir -p $output_dir
fi
path_for_parsed_chains=$output_dir"/parsed_pdbs.jsonl"
python ../helper_scripts/parse_multiple_chains.py --input_path=$folder_with_pdbs --output_path=$path_for_parsed_chains
python ../protein_mpnn_run.py \
--jsonl_path $path_for_parsed_chains \
--out_folder $output_dir \
--num_seq_per_target 2 \
--sampling_temp "0.1" \
--batch_size 1
```
-----------------------------------------------------------------------------------------------------
Example from `vanilla_proteinmpnn/examples/` to design some homomers:
```
folder_with_pdbs="../PDB_homooligomers/pdbs/"
output_dir="../PDB_homooligomers/example_6_outputs"
if [ ! -d $output_dir ]
then
mkdir -p $output_dir
fi
path_for_parsed_chains=$output_dir"/parsed_pdbs.jsonl"
path_for_tied_positions=$output_dir"/tied_pdbs.jsonl"
python ../helper_scripts/parse_multiple_chains.py --input_path=$folder_with_pdbs --output_path=$path_for_parsed_chains
python ../helper_scripts/make_tied_positions_dict.py --input_path=$path_for_parsed_chains --output_path=$path_for_tied_positions --homooligomer 1
python ../protein_mpnn_run.py \
--jsonl_path $path_for_parsed_chains \
--tied_positions_jsonl $path_for_tied_positions \
--out_folder $output_dir \
--num_seq_per_target 2 \
--sampling_temp "0.2" \
--batch_size 1
```
-----------------------------------------------------------------------------------------------------
Example from `vanilla_proteinmpnn/examples/` to design some complexes:
```
folder_with_pdbs="../PDB_complexes/pdbs/"
output_dir="../PDB_complexes/example_4_outputs"
if [ ! -d $output_dir ]
then
mkdir -p $output_dir
fi
path_for_parsed_chains=$output_dir"/parsed_pdbs.jsonl"
path_for_assigned_chains=$output_dir"/assigned_pdbs.jsonl"
path_for_fixed_positions=$output_dir"/fixed_pdbs.jsonl"
chains_to_design="A C"
#The first amino acid in the chain corresponds to 1 and not PDB residues index for now.
fixed_positions="1 2 3 4 5 6 7 8 23 25, 10 11 12 13 14 15 16 17 18 19 20 40" #fixing/not designing residues 1 2 3...25 in chain A and residues 10 11 12...40 in chain C
python ../helper_scripts/parse_multiple_chains.py --input_path=$folder_with_pdbs --output_path=$path_for_parsed_chains
python ../helper_scripts/assign_fixed_chains.py --input_path=$path_for_parsed_chains --output_path=$path_for_assigned_chains --chain_list "$chains_to_design"
python ../helper_scripts/make_fixed_positions_dict.py --input_path=$path_for_parsed_chains --output_path=$path_for_fixed_positions --chain_list "$chains_to_design" --position_list "$fixed_positions"
python ../protein_mpnn_run.py \
--jsonl_path $path_for_parsed_chains \
--chain_id_jsonl $path_for_assigned_chains \
--fixed_positions_jsonl $path_for_fixed_positions \
--out_folder $output_dir \
--num_seq_per_target 2 \
--sampling_temp "0.1" \
--batch_size 1
```
|