Spaces:
Running
on
T4

Running
on
T4
File size: 27,742 Bytes
59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 59a9ccf 8a6c540 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382 383 384 385 386 387 388 389 390 391 392 393 394 395 396 397 398 399 400 401 402 403 404 405 406 407 408 409 410 411 412 413 414 415 416 417 418 419 420 421 422 423 424 425 426 427 428 429 430 431 432 433 434 435 436 437 438 439 440 441 442 443 444 445 446 447 448 449 450 451 452 453 454 455 456 457 458 459 460 461 462 463 464 465 466 467 468 469 470 471 472 473 474 475 476 477 478 479 480 481 482 483 484 485 486 487 488 489 490 491 492 493 494 495 496 497 498 499 500 501 502 503 504 505 506 507 508 509 510 511 512 513 514 515 516 517 518 519 520 521 522 523 524 525 526 527 528 529 530 531 532 533 534 535 536 537 538 539 540 541 542 543 544 545 546 547 548 549 550 551 552 553 554 555 556 557 558 559 560 561 562 563 564 565 566 567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 585 586 587 588 589 590 591 592 593 594 595 596 597 598 599 600 601 602 603 604 605 606 607 |
import os,sys
# install environment goods
#os.system("pip -q install dgl -f https://data.dgl.ai/wheels/cu113/repo.html")
os.system('pip install dgl==1.0.2+cu116 -f https://data.dgl.ai/wheels/cu116/repo.html')
#os.system('pip install gradio')
os.environ["DGLBACKEND"] = "pytorch"
#os.system(f'pip install -r ./PROTEIN_GENERATOR/requirements.txt')
print('Modules installed')
os.system('pip install --force gradio==3.28.3')
os.environ["DGLBACKEND"] = "pytorch"
if not os.path.exists('./SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt'):
print('Downloading model weights 1')
os.system('wget http://files.ipd.uw.edu/pub/sequence_diffusion/checkpoints/SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt')
print('Successfully Downloaded')
if not os.path.exists('./SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt'):
print('Downloading model weights 2')
os.system('wget http://files.ipd.uw.edu/pub/sequence_diffusion/checkpoints/SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt')
print('Successfully Downloaded')
import numpy as np
import gradio as gr
import py3Dmol
from io import StringIO
import json
import secrets
import copy
import matplotlib.pyplot as plt
from utils.sampler import HuggingFace_sampler
plt.rcParams.update({'font.size': 13})
with open('./tmp/args.json','r') as f:
args = json.load(f)
# manually set checkpoint to load
args['checkpoint'] = None
args['dump_trb'] = False
args['dump_args'] = True
args['save_best_plddt'] = True
args['T'] = 25
args['strand_bias'] = 0.0
args['loop_bias'] = 0.0
args['helix_bias'] = 0.0
def protein_diffusion_model(sequence, seq_len, helix_bias, strand_bias, loop_bias,
secondary_structure, aa_bias, aa_bias_potential,
#target_charge, target_ph, charge_potential,
num_steps, noise, hydrophobic_target_score, hydrophobic_potential,
contigs, pssm, seq_mask, str_mask, rewrite_pdb):
dssp_checkpoint = './SEQDIFF_230205_dssp_hotspots_25mask_EQtasks_mod30.pt'
og_checkpoint = './SEQDIFF_221219_equalTASKS_nostrSELFCOND_mod30.pt'
model_args = copy.deepcopy(args)
# make sampler
S = HuggingFace_sampler(args=model_args)
# get random prefix
S.out_prefix = './tmp/'+secrets.token_hex(nbytes=10).upper()
# set args
S.args['checkpoint'] = None
S.args['dump_trb'] = False
S.args['dump_args'] = True
S.args['save_best_plddt'] = True
S.args['T'] = 20
S.args['strand_bias'] = 0.0
S.args['loop_bias'] = 0.0
S.args['helix_bias'] = 0.0
S.args['potentials'] = None
S.args['potential_scale'] = None
S.args['aa_composition'] = None
# get sequence if entered and make sure all chars are valid
alt_aa_dict = {'B':['D','N'],'J':['I','L'],'U':['C'],'Z':['E','Q'],'O':['K']}
if sequence not in ['',None]:
L = len(sequence)
aa_seq = []
for aa in sequence.upper():
if aa in alt_aa_dict.keys():
aa_seq.append(np.random.choice(alt_aa_dict[aa]))
else:
aa_seq.append(aa)
S.args['sequence'] = aa_seq
elif contigs not in ['',None]:
S.args['contigs'] = [contigs]
else:
S.args['contigs'] = [f'{seq_len}']
L = int(seq_len)
print('DEBUG: ',rewrite_pdb)
if rewrite_pdb not in ['',None]:
S.args['pdb'] = rewrite_pdb.name
if seq_mask not in ['',None]:
S.args['inpaint_seq'] = [seq_mask]
if str_mask not in ['',None]:
S.args['inpaint_str'] = [str_mask]
if secondary_structure in ['',None]:
secondary_structure = None
else:
secondary_structure = ''.join(['E' if x == 'S' else x for x in secondary_structure])
if L < len(secondary_structure):
secondary_structure = secondary_structure[:len(sequence)]
elif L == len(secondary_structure):
pass
else:
dseq = L - len(secondary_structure)
secondary_structure += secondary_structure[-1]*dseq
# potentials
potential_list = []
potential_bias_list = []
if aa_bias not in ['',None]:
potential_list.append('aa_bias')
S.args['aa_composition'] = aa_bias
if aa_bias_potential in ['',None]:
aa_bias_potential = 3
potential_bias_list.append(str(aa_bias_potential))
'''
if target_charge not in ['',None]:
potential_list.append('charge')
if charge_potential in ['',None]:
charge_potential = 1
potential_bias_list.append(str(charge_potential))
S.args['target_charge'] = float(target_charge)
if target_ph in ['',None]:
target_ph = 7.4
S.args['target_pH'] = float(target_ph)
'''
if hydrophobic_target_score not in ['',None]:
potential_list.append('hydrophobic')
S.args['hydrophobic_score'] = float(hydrophobic_target_score)
if hydrophobic_potential in ['',None]:
hydrophobic_potential = 3
potential_bias_list.append(str(hydrophobic_potential))
if pssm not in ['',None]:
potential_list.append('PSSM')
potential_bias_list.append('5')
S.args['PSSM'] = pssm.name
if len(potential_list) > 0:
S.args['potentials'] = ','.join(potential_list)
S.args['potential_scale'] = ','.join(potential_bias_list)
# normalise secondary_structure bias from range 0-0.3
S.args['secondary_structure'] = secondary_structure
S.args['helix_bias'] = helix_bias
S.args['strand_bias'] = strand_bias
S.args['loop_bias'] = loop_bias
# set T
if num_steps in ['',None]:
S.args['T'] = 20
else:
S.args['T'] = int(num_steps)
# noise
if 'normal' in noise:
S.args['sample_distribution'] = noise
S.args['sample_distribution_gmm_means'] = [0]
S.args['sample_distribution_gmm_variances'] = [1]
elif 'gmm2' in noise:
S.args['sample_distribution'] = noise
S.args['sample_distribution_gmm_means'] = [-1,1]
S.args['sample_distribution_gmm_variances'] = [1,1]
elif 'gmm3' in noise:
S.args['sample_distribution'] = noise
S.args['sample_distribution_gmm_means'] = [-1,0,1]
S.args['sample_distribution_gmm_variances'] = [1,1,1]
if secondary_structure not in ['',None] or helix_bias+strand_bias+loop_bias > 0:
S.args['checkpoint'] = dssp_checkpoint
S.args['d_t1d'] = 29
print('using dssp checkpoint')
else:
S.args['checkpoint'] = og_checkpoint
S.args['d_t1d'] = 24
print('using og checkpoint')
for k,v in S.args.items():
print(f"{k} --> {v}")
# init S
S.model_init()
S.diffuser_init()
S.setup()
# sampling loop
plddt_data = []
for j in range(S.max_t):
output_seq, output_pdb, plddt = S.take_step_get_outputs(j)
plddt_data.append(plddt)
yield output_seq, output_pdb, display_pdb(output_pdb), get_plddt_plot(plddt_data, S.max_t)
output_seq, output_pdb, plddt = S.get_outputs()
yield output_seq, output_pdb, display_pdb(output_pdb), get_plddt_plot(plddt_data, S.max_t)
def get_plddt_plot(plddt_data, max_t):
x = [i+1 for i in range(len(plddt_data))]
fig, ax = plt.subplots(figsize=(15,6))
ax.plot(x,plddt_data,color='#661dbf', linewidth=3,marker='o')
ax.set_xticks([i+1 for i in range(max_t)])
ax.set_yticks([(i+1)/10 for i in range(10)])
ax.set_ylim([0,1])
ax.set_ylabel('model confidence (plddt)')
ax.set_xlabel('diffusion steps (t)')
return fig
def display_pdb(path_to_pdb):
'''
#function to display pdb in py3dmol
'''
pdb = open(path_to_pdb, "r").read()
view = py3Dmol.view(width=500, height=500)
view.addModel(pdb, "pdb")
view.setStyle({'model': -1}, {"cartoon": {'colorscheme':{'prop':'b','gradient':'roygb','min':0,'max':1}}})#'linear', 'min': 0, 'max': 1, 'colors': ["#ff9ef0","#a903fc",]}}})
view.zoomTo()
output = view._make_html().replace("'", '"')
print(view._make_html())
x = f"""<!DOCTYPE html><html></center> {output} </center></html>""" # do not use ' in this input
return f"""<iframe height="500px" width="100%" name="result" allow="midi; geolocation; microphone; camera;
display-capture; encrypted-media;" sandbox="allow-modals allow-forms
allow-scripts allow-same-origin allow-popups
allow-top-navigation-by-user-activation allow-downloads" allowfullscreen=""
allowpaymentrequest="" frameborder="0" srcdoc='{x}'></iframe>"""
'''
return f"""<iframe style="width: 100%; height:700px" name="result" allow="midi; geolocation; microphone; camera;
display-capture; encrypted-media;" sandbox="allow-modals allow-forms
allow-scripts allow-same-origin allow-popups
allow-top-navigation-by-user-activation allow-downloads" allowfullscreen=""
allowpaymentrequest="" frameborder="0" srcdoc='{x}'></iframe>"""
'''
# MOTIF SCAFFOLDING
def get_motif_preview(pdb_id, contigs):
'''
#function to display selected motif in py3dmol
'''
input_pdb = fetch_pdb(pdb_id=pdb_id.lower())
# rewrite pdb
parse = parse_pdb(input_pdb)
#output_name = './rewrite_'+input_pdb.split('/')[-1]
#writepdb(output_name, torch.tensor(parse_og['xyz']),torch.tensor(parse_og['seq']))
#parse = parse_pdb(output_name)
output_name = input_pdb
pdb = open(output_name, "r").read()
view = py3Dmol.view(width=500, height=500)
view.addModel(pdb, "pdb")
if contigs in ['',0]:
contigs = ['0']
else:
contigs = [contigs]
print('DEBUG: ',contigs)
pdb_map = get_mappings(ContigMap(parse,contigs))
print('DEBUG: ',pdb_map)
print('DEBUG: ',pdb_map['con_ref_idx0'])
roi = [x[1]-1 for x in pdb_map['con_ref_pdb_idx']]
colormap = {0:'#D3D3D3', 1:'#F74CFF'}
colors = {i+1: colormap[1] if i in roi else colormap[0] for i in range(parse['xyz'].shape[0])}
view.setStyle({"cartoon": {"colorscheme": {"prop": "resi", "map": colors}}})
view.zoomTo()
output = view._make_html().replace("'", '"')
print(view._make_html())
x = f"""<!DOCTYPE html><html></center> {output} </center></html>""" # do not use ' in this input
return f"""<iframe height="500px" width="100%" name="result" allow="midi; geolocation; microphone; camera;
display-capture; encrypted-media;" sandbox="allow-modals allow-forms
allow-scripts allow-same-origin allow-popups
allow-top-navigation-by-user-activation allow-downloads" allowfullscreen=""
allowpaymentrequest="" frameborder="0" srcdoc='{x}'></iframe>""", output_name
def fetch_pdb(pdb_id=None):
if pdb_id is None or pdb_id == "":
return None
else:
os.system(f"wget -qnc https://files.rcsb.org/view/{pdb_id}.pdb")
return f"{pdb_id}.pdb"
# MSA AND PSSM GUIDANCE
def save_pssm(file_upload):
filename = file_upload.name
orig_name = file_upload.orig_name
if filename.split('.')[-1] in ['fasta', 'a3m']:
return msa_to_pssm(file_upload)
return filename
def msa_to_pssm(msa_file):
# Define the lookup table for converting amino acids to indices
aa_to_index = {'A': 0, 'R': 1, 'N': 2, 'D': 3, 'C': 4, 'Q': 5, 'E': 6, 'G': 7, 'H': 8, 'I': 9, 'L': 10,
'K': 11, 'M': 12, 'F': 13, 'P': 14, 'S': 15, 'T': 16, 'W': 17, 'Y': 18, 'V': 19, 'X': 20, '-': 21}
# Open the FASTA file and read the sequences
records = list(SeqIO.parse(msa_file.name, "fasta"))
assert len(records) >= 1, "MSA must contain more than one protein sequecne."
first_seq = str(records[0].seq)
aligned_seqs = [first_seq]
# print(aligned_seqs)
# Perform sequence alignment using the Needleman-Wunsch algorithm
aligner = Align.PairwiseAligner()
aligner.open_gap_score = -0.7
aligner.extend_gap_score = -0.3
for record in records[1:]:
alignment = aligner.align(first_seq, str(record.seq))[0]
alignment = alignment.format().split("\n")
al1 = alignment[0]
al2 = alignment[2]
al1_fin = ""
al2_fin = ""
percent_gap = al2.count('-')/ len(al2)
if percent_gap > 0.4:
continue
for i in range(len(al1)):
if al1[i] != '-':
al1_fin += al1[i]
al2_fin += al2[i]
aligned_seqs.append(str(al2_fin))
# Get the length of the aligned sequences
aligned_seq_length = len(first_seq)
# Initialize the position scoring matrix
matrix = np.zeros((22, aligned_seq_length))
# Iterate through the aligned sequences and count the amino acids at each position
for seq in aligned_seqs:
#print(seq)
for i in range(aligned_seq_length):
if i == len(seq):
break
amino_acid = seq[i]
if amino_acid.upper() not in aa_to_index.keys():
continue
else:
aa_index = aa_to_index[amino_acid.upper()]
matrix[aa_index, i] += 1
# Normalize the counts to get the frequency of each amino acid at each position
matrix /= len(aligned_seqs)
print(len(aligned_seqs))
matrix[20:,]=0
outdir = ".".join(msa_file.name.split('.')[:-1]) + ".csv"
np.savetxt(outdir, matrix[:21,:].T, delimiter=",")
return outdir
def get_pssm(fasta_msa, input_pssm):
if input_pssm not in ['',None]:
outdir = input_pssm.name
else:
outdir = save_pssm(fasta_msa)
pssm = np.loadtxt(outdir, delimiter=",", dtype=float)
fig, ax = plt.subplots(figsize=(15,6))
plt.imshow(torch.permute(torch.tensor(pssm),(1,0)))
return fig, outdir
#toggle options
def toggle_seq_input(choice):
if choice == "protein length":
return gr.update(visible=True, value=None), gr.update(visible=False, value=None)
elif choice == "custom sequence":
return gr.update(visible=False, value=None), gr.update(visible=True, value=None)
def toggle_secondary_structure(choice):
if choice == "sliders":
return gr.update(visible=True, value=None),gr.update(visible=True, value=None),gr.update(visible=True, value=None),gr.update(visible=False, value=None)
elif choice == "explicit":
return gr.update(visible=False, value=None),gr.update(visible=False, value=None),gr.update(visible=False, value=None),gr.update(visible=True, value=None)
# Define the Gradio interface
with gr.Blocks(theme='ParityError/Interstellar') as demo:
gr.Markdown(f"""# Protein Generation via Diffusion in Sequence Space""")
with gr.Row():
with gr.Column(min_width=500):
gr.Markdown(f"""
## How does it work?\n
--- [PREPRINT](https://biorxiv.org/content/10.1101/2023.05.08.539766v1) ---
Protein sequence and structure co-generation is a long outstanding problem in the field of protein design. By implementing [ddpm](https://arxiv.org/abs/2006.11239) style diffusion over protein seqeuence space we generate protein sequence and structure pairs. Starting with [RoseTTAFold](https://www.science.org/doi/10.1126/science.abj8754), a protein structure prediction network, we finetuned it to predict sequence and structure given a partially noised sequence. By applying losses to both the predicted sequence and structure the model is forced to generate meaningful pairs. Diffusing in sequence space makes it easy to implement potentials to guide the diffusive process toward particular amino acid composition, net charge, and more! Furthermore, you can sample proteins from a family of sequences or even train a small sequence to function classifier to guide generation toward desired sequences.
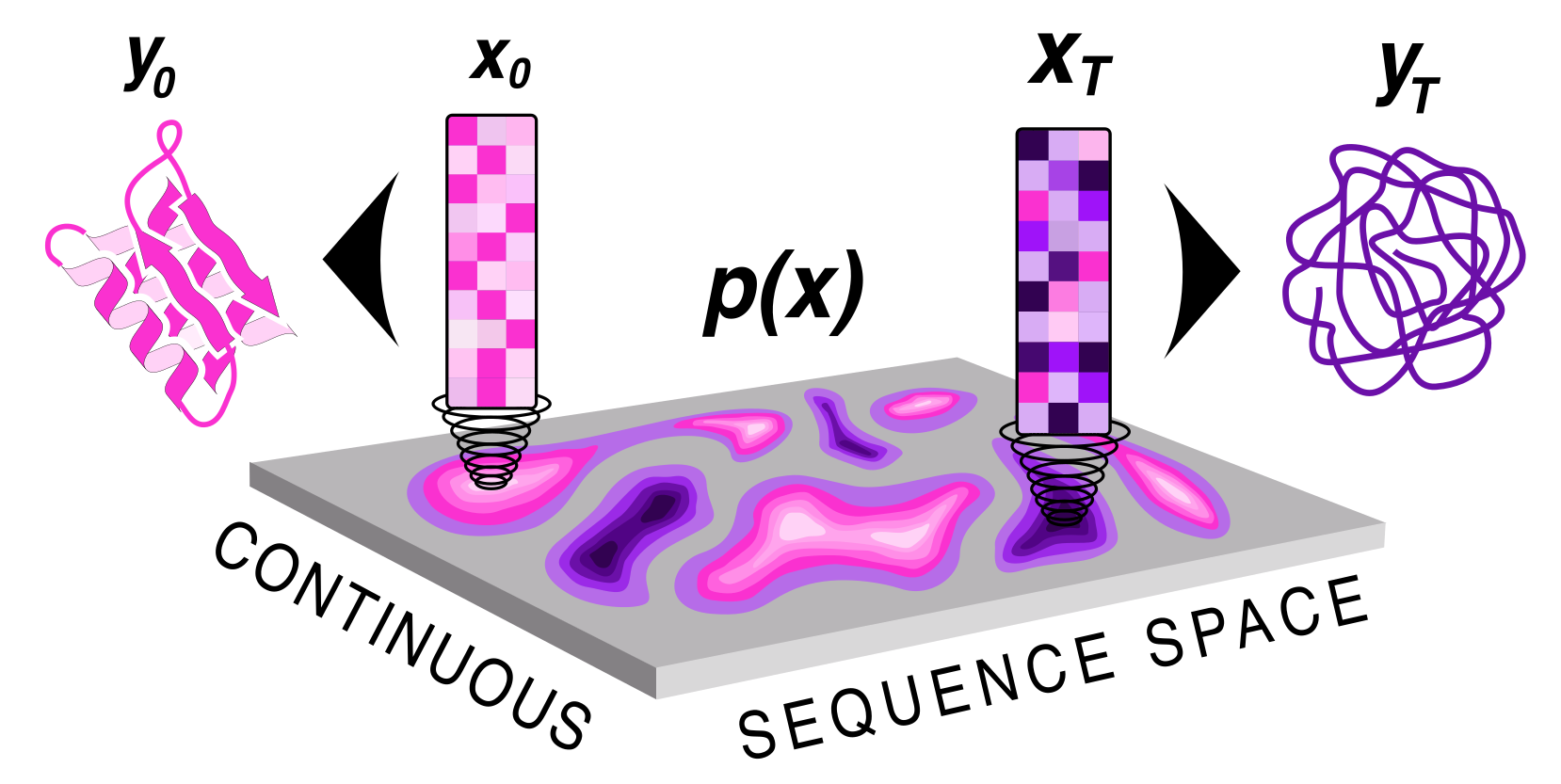
## How to use it?\n
A user can either design a custom input sequence to diffuse from or specify a length below. To scaffold a sequence use the following format where X represent residues to diffuse: XXXXXXXXSCIENCESCIENCEXXXXXXXXXXXXXXXXXXX. You can even design a protein with your name XXXXXXXXXXXXNAMEHEREXXXXXXXXXXXXX!
### Acknowledgements\n
Thank you to Simon Dürr and the Hugging Face team for setting us up with a community GPU grant!
""")
gr.Markdown("""
## Model in Action

""")
with gr.Row().style(equal_height=False):
with gr.Column():
with gr.Tabs():
with gr.TabItem("Inputs"):
gr.Markdown("""## INPUTS""")
gr.Markdown("""#### Start Sequence
Specify the protein length for complete unconditional generation, or scaffold a motif (or your name) using the custom sequence input""")
seq_opt = gr.Radio(["protein length","custom sequence"], label="How would you like to specify the starting sequence?", value='protein length')
sequence = gr.Textbox(label="custom sequence", lines=1, placeholder='AMINO ACIDS: A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y\n MASK TOKEN: X', visible=False)
seq_len = gr.Slider(minimum=5.0, maximum=250.0, label="protein length", value=100, visible=True)
seq_opt.change(fn=toggle_seq_input,
inputs=[seq_opt],
outputs=[seq_len, sequence],
queue=False)
gr.Markdown("""### Optional Parameters""")
with gr.Accordion(label='Secondary Structure',open=True):
gr.Markdown("""Try changing the sliders or inputing explicit secondary structure conditioning for each residue""")
sec_str_opt = gr.Radio(["sliders","explicit"], label="How would you like to specify secondary structure?", value='sliders')
secondary_structure = gr.Textbox(label="secondary structure", lines=1, placeholder='HELIX = H STRAND = S LOOP = L MASK = X(must be the same length as input sequence)', visible=False)
with gr.Column():
helix_bias = gr.Slider(minimum=0.0, maximum=0.05, label="helix bias", visible=True)
strand_bias = gr.Slider(minimum=0.0, maximum=0.05, label="strand bias", visible=True)
loop_bias = gr.Slider(minimum=0.0, maximum=0.20, label="loop bias", visible=True)
sec_str_opt.change(fn=toggle_secondary_structure,
inputs=[sec_str_opt],
outputs=[helix_bias,strand_bias,loop_bias,secondary_structure],
queue=False)
with gr.Accordion(label='Amino Acid Compositional Bias',open=False):
gr.Markdown("""Bias sequence composition for particular amino acids by specifying the one letter code followed by the fraction to bias. This can be input as a list for example: W0.2,E0.1""")
with gr.Row():
aa_bias = gr.Textbox(label="aa bias", lines=1, placeholder='specify one letter AA and fraction to bias, for example W0.1 or M0.1,K0.1' )
aa_bias_potential = gr.Textbox(label="aa bias scale", lines=1, placeholder='AA Bias potential scale (recomended range 1.0-5.0)')
'''
with gr.Accordion(label='Charge Bias',open=False):
gr.Markdown("""Bias for a specified net charge at a particular pH using the boxes below""")
with gr.Row():
target_charge = gr.Textbox(label="net charge", lines=1, placeholder='net charge to target')
target_ph = gr.Textbox(label="pH", lines=1, placeholder='pH at which net charge is desired')
charge_potential = gr.Textbox(label="charge potential scale", lines=1, placeholder='charge potential scale (recomended range 1.0-5.0)')
'''
with gr.Accordion(label='Hydrophobic Bias',open=False):
gr.Markdown("""Bias for or against hydrophobic composition, to get more soluble proteins, bias away with a negative target score (ex. -5)""")
with gr.Row():
hydrophobic_target_score = gr.Textbox(label="hydrophobic score", lines=1, placeholder='hydrophobic score to target (negative score is good for solublility)')
hydrophobic_potential = gr.Textbox(label="hydrophobic potential scale", lines=1, placeholder='hydrophobic potential scale (recomended range 1.0-2.0)')
with gr.Accordion(label='Diffusion Params',open=False):
gr.Markdown("""Increasing T to more steps can be helpful for harder design challenges, sampling from different distributions can change the sequence and structural composition""")
with gr.Row():
num_steps = gr.Textbox(label="T", lines=1, placeholder='number of diffusion steps (25 or less will speed things up)')
noise = gr.Dropdown(['normal','gmm2 [-1,1]','gmm3 [-1,0,1]'], label='noise type', value='normal')
with gr.TabItem("Motif Selection"):
gr.Markdown("""### Motif Selection Preview""")
gr.Markdown('Contigs explained: to grab residues (seq and str) on a pdb chain you will provide the chain letter followed by a range of residues as indexed in the pdb file for example (A3-10) is the syntax to select residues 3-10 on chain A (the chain always needs to be specified). To add diffused residues to either side of this motif you can specify a range or discrete value without a chain letter infront. To add 15 residues before the motif and 20-30 residues (randomly sampled) after use the following syntax: 15,A3-10,20-30 commas are used to separate regions selected from the pdb and designed (diffused) resiudes which will be added. ')
pdb_id_code = gr.Textbox(label="PDB ID", lines=1, placeholder='INPUT PDB ID TO FETCH (ex. 1DPX)', visible=True)
contigs = gr.Textbox(label="contigs", lines=1, placeholder='specify contigs to grab particular residues from pdb ()', visible=True)
gr.Markdown('Using the same contig syntax, seq or str of input motif residues can be masked, allowing the model to hold strucutre fixed and design sequence or vice-versa')
with gr.Row():
seq_mask = gr.Textbox(label='seq mask',lines=1,placeholder='input residues to mask sequence')
str_mask = gr.Textbox(label='str mask',lines=1,placeholder='input residues to mask structure')
preview_viewer = gr.HTML()
rewrite_pdb = gr.File(label='PDB file')
preview_btn = gr.Button("Preview Motif")
with gr.TabItem("MSA to PSSM"):
gr.Markdown("""### MSA to PSSM Generation""")
gr.Markdown('input either an MSA or PSSM to guide the model toward generating samples within your family of interest')
with gr.Row():
fasta_msa = gr.File(label='MSA')
input_pssm = gr.File(label='PSSM (.csv)')
pssm = gr.File(label='Generated PSSM')
pssm_view = gr.Plot(label='PSSM Viewer')
pssm_gen_btn = gr.Button("Generate PSSM")
btn = gr.Button("GENERATE")
#with gr.Row():
with gr.Column():
gr.Markdown("""## OUTPUTS""")
gr.Markdown("""#### Confidence score for generated structure at each timestep""")
plddt_plot = gr.Plot(label='plddt at step t')
gr.Markdown("""#### Output protein sequnece""")
output_seq = gr.Textbox(label="sequence")
gr.Markdown("""#### Download PDB file""")
output_pdb = gr.File(label="PDB file")
gr.Markdown("""#### Structure viewer""")
output_viewer = gr.HTML()
gr.Markdown("""### Don't know where to get started? Click on an example below to try it out!""")
gr.Examples(
[["","125",0.0,0.0,0.2,"","","","20","normal",'','','',None,'','',None],
["","100",0.0,0.0,0.0,"","W0.2","2","20","normal",'','','',None,'','',None],
["","100",0.0,0.0,0.0,
"XXHHHHHHHHHXXXXXXXHHHHHHHHHXXXXXXXHHHHHHHHXXXXSSSSSSSSSSSXXXXXXXXSSSSSSSSSSSSXXXXXXXSSSSSSSSSXXXXXXX",
"","","25","normal",'','','',None,'','',None],
["XXXXXXXXXXXXXXXXXXXXXXXXXIPDXXXXXXXXXXXXXXXXXXXXXXPEPSEQXXXXXXXXXXXXXXXXXXXXXXXXXXIPDXXXXXXXXXXXXXXXXXXX",
"",0.0,0.0,0.0,"","","","25","normal",'','','',None,'','',None],
["","",0.0,0.0,0.0,"","","","25","normal",'','',
'9,D10-11,8,D20-20,4,D25-35,65,D101-101,2,D104-105,8,D114-116,15,D132-138,6,D145-145,2,D148-148,12,D161-161,3',
'./tmp/PSSM_lysozyme.csv',
'D25-25,D27-31,D33-35,D132-137',
'D26-26','./tmp/150l.pdb']
],
inputs=[sequence,
seq_len,
helix_bias,
strand_bias,
loop_bias,
secondary_structure,
aa_bias,
aa_bias_potential,
#target_charge,
#target_ph,
#charge_potential,
num_steps,
noise,
hydrophobic_target_score,
hydrophobic_potential,
contigs,
pssm,
seq_mask,
str_mask,
rewrite_pdb],
outputs=[output_seq,
output_pdb,
output_viewer,
plddt_plot],
fn=protein_diffusion_model,
)
preview_btn.click(get_motif_preview,[pdb_id_code, contigs],[preview_viewer, rewrite_pdb])
pssm_gen_btn.click(get_pssm,[fasta_msa,input_pssm],[pssm_view, pssm])
btn.click(protein_diffusion_model,
[sequence,
seq_len,
helix_bias,
strand_bias,
loop_bias,
secondary_structure,
aa_bias,
aa_bias_potential,
#target_charge,
#target_ph,
#charge_potential,
num_steps,
noise,
hydrophobic_target_score,
hydrophobic_potential,
contigs,
pssm,
seq_mask,
str_mask,
rewrite_pdb],
[output_seq,
output_pdb,
output_viewer,
plddt_plot])
demo.queue()
demo.launch(debug=True)
|