Spaces:
Sleeping

Sleeping
| #!/usr/bin/env python | |
| # coding: utf-8 | |
| # # Celltype auto annotation with MetaTiME | |
| # | |
| # MetaTiME learns data-driven, interpretable, and reproducible gene programs by integrating millions of single cells from hundreds of tumor scRNA-seq data. The idea is to learn a map of single-cell space with biologically meaningful directions from large-scale data, which helps understand functional cell states and transfers knowledge to new data analysis. MetaTiME provides pretrained meta-components (MeCs) to automatically annotate fine-grained cell states and plot signature continuum for new single-cells of tumor microenvironment. | |
| # | |
| # Here, we integrate MetaTiME in omicverse. This tutorial demonstrates how to use [MetaTiME (original code)](https://github.com/yi-zhang/MetaTiME/blob/main/docs/notebooks/metatime_annotator.ipynb) to annotate celltype in TME | |
| # | |
| # Paper: [MetaTiME integrates single-cell gene expression to characterize the meta-components of the tumor immune microenvironment](https://www.nature.com/articles/s41467-023-38333-8) | |
| # | |
| # Code: https://github.com/yi-zhang/MetaTiME | |
| # | |
| # Colab_Reproducibility:https://colab.research.google.com/drive/1isvjTfSFM2cy6GzHWAwbuvSjveEJijzP?usp=sharing | |
| # | |
| # 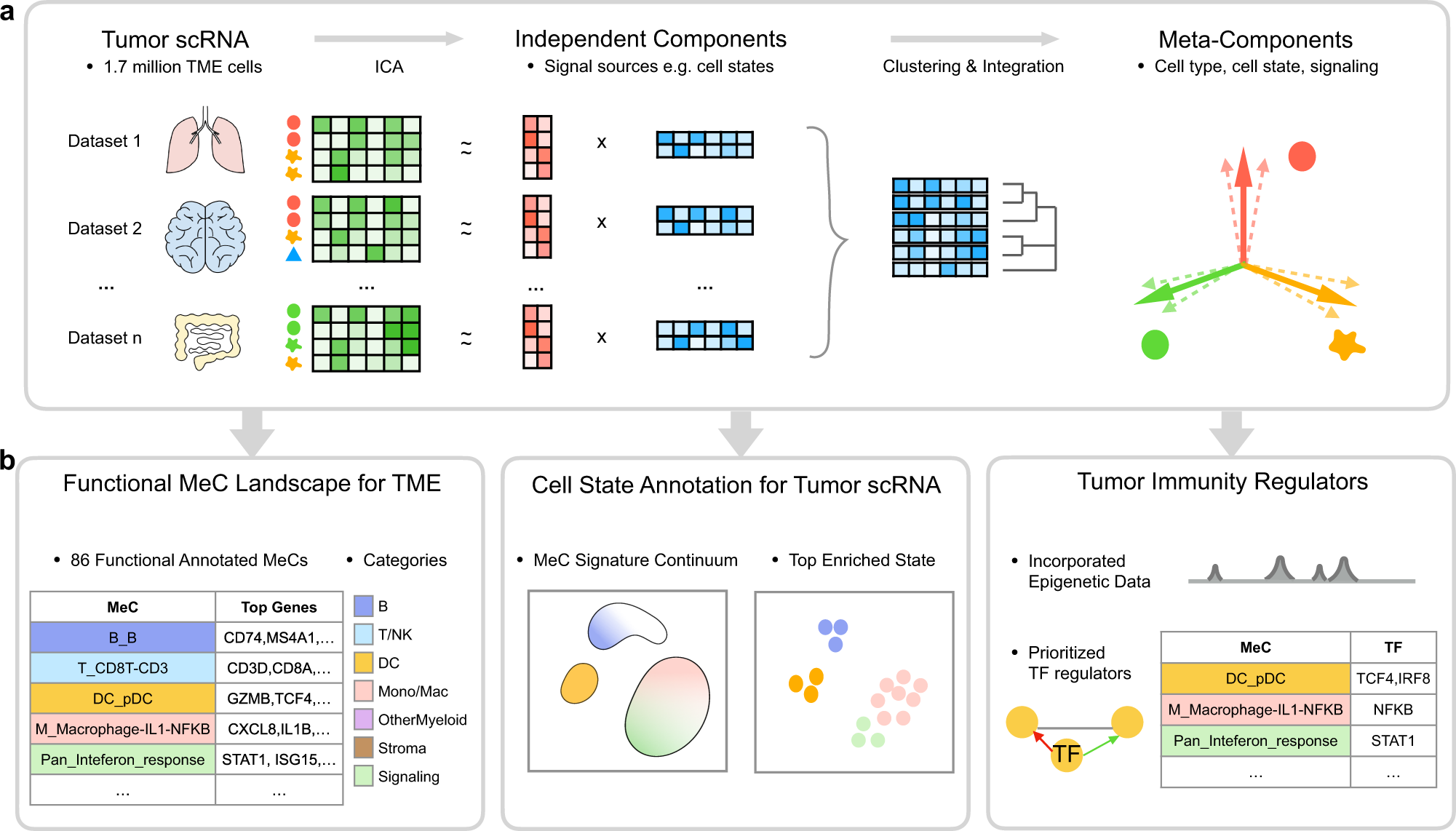 | |
| # In[1]: | |
| import omicverse as ov | |
| ov.utils.ov_plot_set() | |
| # ## Data normalize and Batch remove | |
| # | |
| # The sample data has multiple patients , and we can use batch correction on patients. Here, we using [scVI](https://docs.scvi-tools.org/en/stable/) to remove batch. | |
| # | |
| # <div class="admonition warning"> | |
| # <p class="admonition-title">Note</p> | |
| # <p> | |
| # If your data contains count matrix, we provide a wrapped function for pre-processing the data. Otherwise, if the data is already depth-normalized, log-transformed, and cells are filtered, we can skip this step. | |
| # </p> | |
| # </div> | |
| # In[ ]: | |
| ''' | |
| import scvi | |
| scvi.model.SCVI.setup_anndata(adata, layer="counts", batch_key="patient") | |
| vae = scvi.model.SCVI(adata, n_layers=2, n_latent=30, gene_likelihood="nb") | |
| vae.train() | |
| adata.obsm["X_scVI"] = vae.get_latent_representation() | |
| ''' | |
| # Example data can be obtained from figshare: https://figshare.com/ndownloader/files/41440050 | |
| # In[2]: | |
| import scanpy as sc | |
| adata=sc.read('TiME_adata_scvi.h5ad') | |
| adata | |
| # It is recommended that malignant cells are identified first and removed for best practice in cell state annotation. | |
| # | |
| # In the BCC data, the cluster of malignant cells are identified with `inferCNV`. We can use the pre-saved column 'isTME' to keep Tumor Microenvironment cells. | |
| # | |
| # These are the authors' exact words, but tests have found that the difference in annotation effect is not that great even without removing the malignant cells | |
| # | |
| # But I think this step is not necessary | |
| # In[3]: | |
| #adata = adata[adata.obs['isTME']] | |
| # ## Neighborhood graph calculated | |
| # | |
| # We note that scVI was used earlier to remove the batch effect from the data, so we need to recalculate the neighbourhood map based on what is stored in `adata.obsm['X_scVI']`. Note that if you are not using scVI but using another method to calculate the neighbourhood map, such as `X_pca`, then you need to change `X_scVI` to `X_pca` to complete the calculation | |
| # | |
| # ``` | |
| # #Example | |
| # #sc.tl.pca(adata) | |
| # #sc.pp.neighbors(adata, use_rep="X_pca") | |
| # ``` | |
| # In[4]: | |
| sc.pp.neighbors(adata, use_rep="X_scVI") | |
| # To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated. | |
| # In[5]: | |
| adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["X_scVI"]) | |
| # In[6]: | |
| sc.pl.embedding( | |
| adata, | |
| basis="X_mde", | |
| color=["patient"], | |
| frameon=False, | |
| ncols=1, | |
| ) | |
| # In[7]: | |
| #adata.write_h5ad('adata_mde.h5ad',compression='gzip') | |
| #adata=sc.read('adata_mde.h5ad') | |
| # ## MeteTiME model init | |
| # | |
| # Next, let's load the pre-computed MetaTiME MetaComponents (MeCs), and their functional annotation. | |
| # In[8]: | |
| TiME_object=ov.single.MetaTiME(adata,mode='table') | |
| # We can over-cluster the cells which is useful for fine-grained cell state annotation. | |
| # | |
| # As the resolution gets larger, the number of clusters gets larger | |
| # In[9]: | |
| TiME_object.overcluster(resolution=8,clustercol = 'overcluster',) | |
| # ## TME celltype predicted | |
| # | |
| # We using `TiME_object.predictTiME()` to predicted the latent celltype in TME. | |
| # | |
| # - The minor celltype will be stored in `adata.obs['MetaTiME']` | |
| # - The major celltype will be stored in `adata.obs['Major_MetaTiME']` | |
| # In[10]: | |
| TiME_object.predictTiME(save_obs_name='MetaTiME') | |
| # ## Visualize | |
| # | |
| # The original author provides a drawing function that effectively avoids overlapping labels. Here I have expanded its parameters so that it can be visualised using parameters other than X_umap | |
| # In[13]: | |
| fig,ax=TiME_object.plot(cluster_key='MetaTiME',basis='X_mde',dpi=80) | |
| #fig.save | |
| # We can also use `sc.pl.embedding` to visualize the celltype | |
| # In[15]: | |
| sc.pl.embedding( | |
| adata, | |
| basis="X_mde", | |
| color=["Major_MetaTiME"], | |
| frameon=False, | |
| ncols=1, | |
| ) | |
| # In[ ]: | |