Spaces:
Sleeping

Sleeping
| #!/usr/bin/env python | |
| # coding: utf-8 | |
| # # Mapping single-cell profile onto spatial profile | |
| # | |
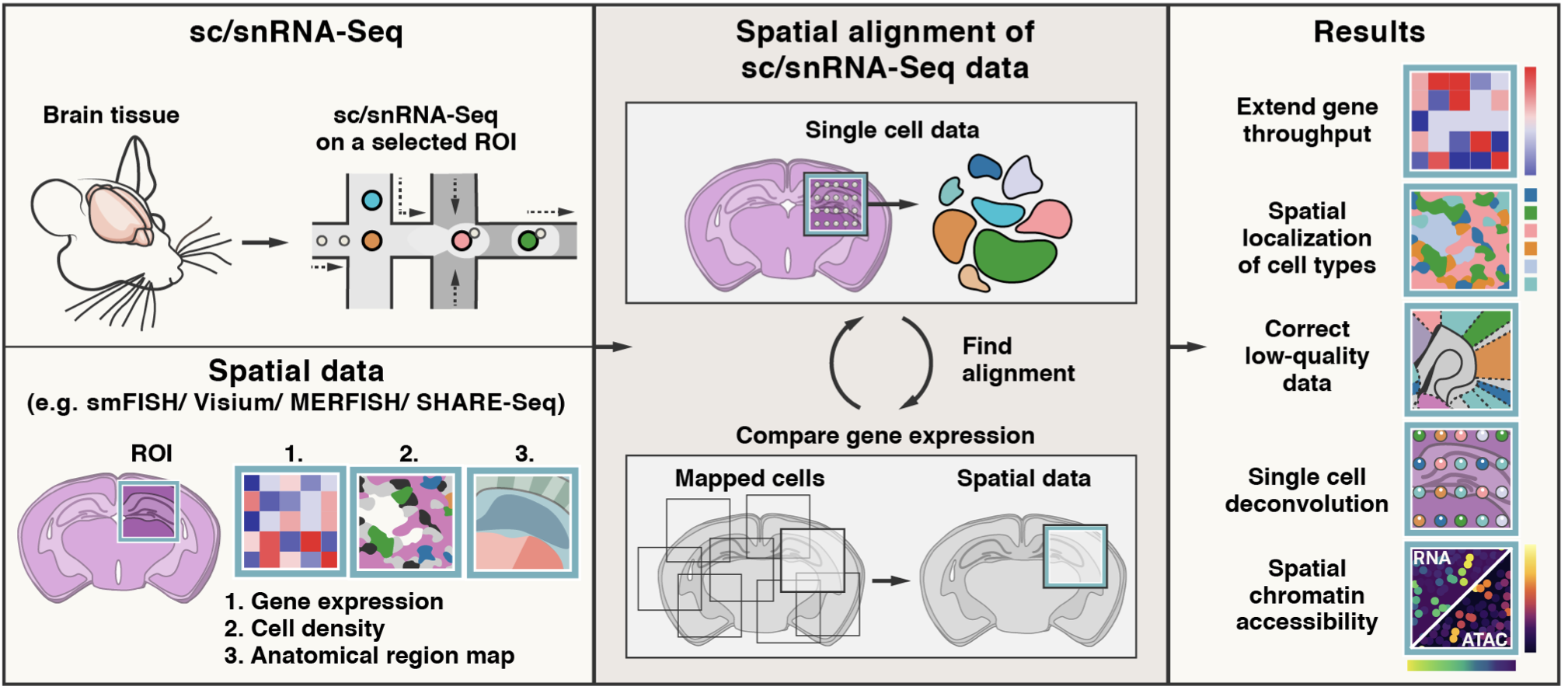
| # Tangram is a method for mapping single-cell (or single-nucleus) gene expression data onto spatial gene expression data. Tangram takes as input a single-cell dataset and a spatial dataset, collected from the same anatomical region/tissue type. Via integration, Tangram creates new spatial data by aligning the scRNAseq profiles in space. This allows to project every annotation in the scRNAseq (e.g. cell types, program usage) on space. | |
| # | |
| # The most common application of Tangram is to resolve cell types in space. Another usage is to correct gene expression from spatial data: as scRNA-seq data are less prone to dropout than (e.g.) Visium or Slide-seq, the “new” spatial data generated by Tangram resolve many more genes. As a result, we can visualize program usage in space, which can be used for ligand-receptor pair discovery or, more generally, cell-cell communication mechanisms. If cell segmentation is available, Tangram can be also used for deconvolution of spatial data. If your single cell are multimodal, Tangram can be used to spatially resolve other modalities, such as chromatin accessibility. | |
| # | |
| # Biancalani, T., Scalia, G., Buffoni, L. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat Methods 18, 1352–1362 (2021). https://doi.org/10.1038/s41592-021-01264-7 | |
| # | |
| #  | |
| # In[1]: | |
| import omicverse as ov | |
| #print(f"omicverse version: {ov.__version__}") | |
| import scanpy as sc | |
| #print(f"scanpy version: {sc.__version__}") | |
| ov.utils.ov_plot_set() | |
| # ## Prepared scRNA-seq | |
| # | |
| # Published scRNA-seq datasets of lymph nodes have typically lacked an adequate representation of germinal centre-associated immune cell populations due to age of patient donors. We, therefore, include scRNA-seq datasets spanning lymph nodes, spleen and tonsils in our single-cell reference to ensure that we captured the full diversity of immune cell states likely to exist in the spatial transcriptomic dataset. | |
| # | |
| # Here we download this dataset, import into anndata and change variable names to ENSEMBL gene identifiers. | |
| # | |
| # Link: https://cell2location.cog.sanger.ac.uk/paper/integrated_lymphoid_organ_scrna/RegressionNBV4Torch_57covariates_73260cells_10237genes/sc.h5ad | |
| # In[2]: | |
| adata_sc=ov.read('data/sc.h5ad') | |
| import matplotlib.pyplot as plt | |
| fig, ax = plt.subplots(figsize=(3,3)) | |
| ov.utils.embedding( | |
| adata_sc, | |
| basis="X_umap", | |
| color=['Subset'], | |
| title='Subset', | |
| frameon='small', | |
| #ncols=1, | |
| wspace=0.65, | |
| #palette=ov.utils.pyomic_palette()[11:], | |
| show=False, | |
| ax=ax | |
| ) | |
| # For data quality control and preprocessing, we can easily use omicverse's own preprocessing functions to do so | |
| # In[3]: | |
| print("RAW",adata_sc.X.max()) | |
| adata_sc=ov.pp.preprocess(adata_sc,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4) | |
| adata_sc.raw = adata_sc | |
| adata_sc = adata_sc[:, adata_sc.var.highly_variable_features] | |
| print("Normalize",adata_sc.X.max()) | |
| # ## Prepared stRNA-seq | |
| # | |
| # First let’s read spatial Visium data from 10X Space Ranger output. Here we use lymph node data generated by 10X and presented in [Kleshchevnikov et al (section 4, Fig 4)](https://www.biorxiv.org/content/10.1101/2020.11.15.378125v1). This dataset can be conveniently downloaded and imported using scanpy. See [this tutorial](https://cell2location.readthedocs.io/en/latest/notebooks/cell2location_short_demo.html) for a more extensive and practical example of data loading (multiple visium samples). | |
| # In[5]: | |
| adata = sc.datasets.visium_sge(sample_id="V1_Human_Lymph_Node") | |
| adata.obs['sample'] = list(adata.uns['spatial'].keys())[0] | |
| adata.var_names_make_unique() | |
| # We used the same pre-processing steps as for scRNA-seq | |
| # | |
| # <div class="admonition warning"> | |
| # <p class="admonition-title">Note</p> | |
| # <p> | |
| # We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code. | |
| # </p> | |
| # </div> | |
| # | |
| # ```python | |
| # #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4) | |
| # #adata.raw = adata | |
| # #adata = adata[:, adata.var.highly_variable_features] | |
| # ``` | |
| # In[6]: | |
| sc.pp.calculate_qc_metrics(adata, inplace=True) | |
| adata = adata[:,adata.var['total_counts']>100] | |
| adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",) | |
| adata.raw = adata | |
| adata = adata[:, adata.var.space_variable_features] | |
| adata_sp=adata.copy() | |
| adata_sp | |
| # ## Tangram model | |
| # | |
| # Tangram is a Python package, written in PyTorch and based on scanpy, for mapping single-cell (or single-nucleus) gene expression data onto spatial gene expression data. The single-cell dataset and the spatial dataset should be collected from the same anatomical region/tissue type, ideally from a biological replicate, and need to share a set of genes. | |
| # | |
| # We can use `omicverse.space.Tangram` to apply the Tangram model. | |
| # In[7]: | |
| tg=ov.space.Tangram(adata_sc,adata_sp,clusters='Subset') | |
| # The function maps iteratively as specified by num_epochs. We typically interrupt mapping after the score plateaus. | |
| # - The score measures the similarity between the gene expression of the mapped cells vs spatial data on the training genes. | |
| # - The default mapping mode is mode=`cells`, which is recommended to run on a GPU. | |
| # - Alternatively, one can specify mode=`clusters` which averages the single cells beloning to the same cluster (pass annotations via cluster_label). This is faster, and is our chioce when scRNAseq and spatial data come from different specimens. | |
| # - If you wish to run Tangram with a GPU, set device=`cuda:0` otherwise use the set device=`cpu`. | |
| # - density_prior specifies the cell density within each spatial voxel. Use uniform if the spatial voxels are at single cell resolution (ie MERFISH). The default value, rna_count_based, assumes that cell density is proportional to the number of RNA molecules | |
| # In[8]: | |
| tg.train(mode="clusters",num_epochs=500,device="cuda:0") | |
| # We can use `tg.cell2location()` to get the cell location in spatial spots. | |
| # In[9]: | |
| adata_plot=tg.cell2location() | |
| adata_plot.obs.columns | |
| # In[10]: | |
| annotation_list=['B_Cycling', 'B_GC_LZ', 'T_CD4+_TfH_GC', 'FDC', | |
| 'B_naive', 'T_CD4+_naive', 'B_plasma', 'Endo'] | |
| sc.pl.spatial(adata_plot, cmap='magma', | |
| # show first 8 cell types | |
| color=annotation_list, | |
| ncols=4, size=1.3, | |
| img_key='hires', | |
| # limit color scale at 99.2% quantile of cell abundance | |
| #vmin=0, vmax='p99.2' | |
| ) | |
| # In[11]: | |
| color_dict=dict(zip(adata_sc.obs['Subset'].cat.categories, | |
| adata_sc.uns['Subset_colors'])) | |
| # In[21]: | |
| import matplotlib as mpl | |
| clust_labels = annotation_list[:5] | |
| clust_col = ['' + str(i) for i in clust_labels] # in case column names differ from labels | |
| with mpl.rc_context({'figure.figsize': (8, 8),'axes.grid': False}): | |
| fig = ov.pl.plot_spatial( | |
| adata=adata_plot, | |
| # labels to show on a plot | |
| color=clust_col, labels=clust_labels, | |
| show_img=True, | |
| # 'fast' (white background) or 'dark_background' | |
| style='fast', | |
| # limit color scale at 99.2% quantile of cell abundance | |
| max_color_quantile=0.992, | |
| # size of locations (adjust depending on figure size) | |
| circle_diameter=3, | |
| reorder_cmap = [#0, | |
| 1,2,3,4,6], #['yellow', 'orange', 'blue', 'green', 'purple', 'grey', 'white'], | |
| colorbar_position='right', | |
| palette=color_dict | |
| ) | |
| # In[ ]: | |