
text
stringlengths 35
445k
| metadata
dict |
|---|---|
# biopython_notebook_1.ipynb
Repository: Deya-B/Bioinformatics-notes
<code>
from Bio.Seq import Seq
seq = Seq('GATTACA')
#Seq methods represent biological sequences as strings
print(seq)
</code>
<code>
seq = Seq('CAT')
for base in seq:
print(base, end=' ')
seq + 'GAT'
</code>
<code>
dna = Seq('GATTACA')
print(dna.complement())
print(dna.reverse_complement())
dna.lower()
</code>
<code>
cds = Seq('GTGTTTTTGGTGTGGTGA')
mRNA = cds.transcribe() #devuelve ARN
print(mRNA)
print(mRNA.back_transcribe())
</code>
<code>
mRNA = Seq('UUGUUUUUGGUGUGGUGA')
############## DIFERENTES TABLAS DE TRANSLACION #########################
print("Translate using standard table: ", mRNA.translate())
print("Translate until stop codon : ", mRNA.translate(to_stop=True)) #tener en cuenta el codon de parada
print("Using a different table : ", mRNA.translate(table=2))
print("by name : ", mRNA.translate(table="Vertebrate Mitochondrial"))
# This next line raises an error because the sequence is not a CDS for table=2
#print("CDS-like RNA : ", mRNA.translate(table=2, cds=True))
# But it is a CDS-like sequence for table=11 (Bacterial)
print("CDS-like RNA : ", mRNA.translate(table=11, cds=True)) #cds es que es una region
#codificante completa
cds = Seq('GTGTTTTTGGTGTGGTGA')
prot = cds.translate(table=11, cds=True)
prot = cds.translate()
prot
</code>
<code>
from Bio.Data import CodonTable
bacterial_table = CodonTable.unambiguous_dna_by_name['Bacterial']
print(bacterial_table)
#como esto sera la salida de nuestra tabla
</code>
<code>
from Bio.SeqRecord import SeqRecord
##### REGISTRO DE SECUENCIA ##### Toda la información de la secuencia + secuencia
sr = SeqRecord(Seq('AAA'), id='1', description='Simple seq', annotations={"molecule_type": "DNA"})
print(sr)
</code>
<code>
from Bio import SeqIO #IO input-output
# Para secuencias pequeñas
record = SeqIO.read("phix174/phix.fa", "fasta") #nombre de fichero, tipo de fichero
print(record)
</code>
<code>
#Para leer archivos mas grandes
for record in SeqIO.parse("other/ls_orchid.fasta", "fasta"): #parametros= nombre, tipo fichero
print(record) #es un iterador, por cada paso nos da cada secuencia
print()
</code>
<code>
for record in SeqIO.parse("other/ls_orchid.gbk", "genbank"):
print(record)
# Nos salen anotaciones, articulis cientificos, gran parte de toda la info que hay
</code>
<code>
iterator = SeqIO.parse("other/ls_orchid.gbk", "genbank")
records_dict = SeqIO.to_dict(iterator) #crear un diccionario
print(records_dict['Z78533.1']) #para imprimir la secuencia con el identificador dado
</code>
<code>
#dd = {}
#dd[record.id]=record #esto metido dentro del bucle te da todos los records
#dd['Z78533.1'] #te daría los detalles de este id solo
#claves en memoria, datos en el disco, hace que vaya todo mas rapido
#importante records_dict.close() al final, porque sino mantiene el fichero abierto
records_dict = SeqIO.index("other/ls_orchid.gbk", "genbank")
print(records_dict['Z78533.1'])
records_dict.close()
</code>
<code>
#para leer ficheros comprimidos
import gzip
with gzip.open("arabidopsis_thaliana/GCF_000001735.3_TAIR10_rna.fna.gz", "rt") as f:
total_len = 0
for sr in SeqIO.parse(f, "fasta"):
total_len += len(sr.seq)
print(total_len)
</code>
<code>
#OCUPA POCO EN MEMORIA Y POCO EN DISCO, es una forma de comprimir en bloques
#pero requiere un formato especial:
# Use this line in a shell to create the bgzf file
# You must have tabix or SAMtools installed
# bgzip -c GCF_000027305.1_ASM2730v1_genomic.gbff > GCF_000027305.1_ASM2730v1_genomic.gbff.bgz
records_dict = SeqIO.index("haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff.bgz", "genbank")
seq = records_dict['NC_000907.1']
records_dict.close()
</code>
<code>
records_dict = SeqIO.index("other/ls_orchid.gbk", "genbank")
record = records_dict['Z78533.1']
records_dict.close()
print("ID:",record.id, "Name:", record.name) #SeqRecord tiene muchas cosas
print("Description:",record.description)
print(record.annotations)
print(record.features)
</code>
<code>
print(record.annotations.keys()) #referencia a las anotaciones
</code>
<code>
for ref in record.annotations['references']:
print(ref)
for l in ref.location:
print(l)
</code>
<code>
from Bio import SeqFeature
records_dict = SeqIO.index("other/ls_orchid.gbk", "genbank")
print(len(records_dict['Z78533.1'].features), "features found")
for feature in records_dict['Z78533.1'].features:
print("----------------------------------------")
print(feature)
records_dict.close()
</code>
<code>
#Leer secuencias de este fichero
file_iter = SeqIO.parse('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.fna', 'fasta')
n_features = 0
#para cada secuencia cogemos las caracteristicas
for seqrecord in file_iter:
n_features += len(seqrecord.features)
print(n_features, "features found")
#esto da 0 porque un fasta no tiene caracteristicas
</code>
<code>
file_iter = SeqIO.parse('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
#file_iter = SeqIO.parse('plamodium_falciparum/GCF_000002765.3_ASM276v1_genomic.gbff', 'genbank')
n_features = 0
for seqrecord in file_iter:
n_features += len(seqrecord.features)
print(n_features, "features found")
#aqui si podemos leer las caracteristicas
</code>
<code>
record = SeqIO.read('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
position = 10000
for feature in record.features:
if position in feature:
print(feature)
print('-----------------------')
</code>
<code>
d = SeqIO.index('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
record = d['NC_000907.1'] # retreieve cromosom
position = 1000 # Position of interest inside the cromosome
# printing publications that studied that cromosome and position
for ref in record.annotations['references']:
if any([position in loc for loc in ref.location]):
print(ref)
</code>
<code>
n_cds = 0
file_iter = SeqIO.parse('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
#file_iter = SeqIO.parse('plamodium_falciparum/GCF_000002765.3_ASM276v1_genomic.gbff', 'genbank')
for seqrec in file_iter:
for feature in seqrec.features:
if feature.type == 'CDS': #leemos el CDS
n_cds += 1
mRNA = feature.extract(seqrec) # para esta seq extraemos el RNA
try:
transl_table = 11
if 'transl_table' in feature.qualifiers:
transl_table = int(feature.qualifiers['transl_table'][0])
p = mRNA.seq.translate(table=transl_table, cds=True)
except:
print("Protein {0} in gene {1} could not be translated!".
format(feature.qualifiers['protein_id'][0], seqrec.id))
print(n_cds)
</code>
<code>
record = SeqIO.read('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
print(record.id)
print("# of feature: ", len(record.features))
print("# of annotations: ", len(record.annotations))
print(record.features[1200])
sub_record = record[612623:620000]
print(sub_record.id)
print("# of feature: ", len(sub_record.features))
print("# of annotations: ", len(sub_record.annotations))
print(sub_record.features[0])
</code>
<code>
n_cds = 0
file_iter = SeqIO.parse('haemophilus_influenzae/GCF_000027305.1_ASM2730v1_genomic.gbff', 'genbank')
#file_iter = SeqIO.parse('plamodium_falciparum/GCF_000002765.3_ASM276v1_genomic.gbff', 'genbank')
for seqrec in file_iter:
for feature in seqrec.features:
if feature.type == 'CDS': #leemos el CDS
n_cds += 1
mRNA = feature.extract(seqrec) # para esta seq extraemos el RNA
try:
transl_table = 1
if 'transl_table' in feature.qualifiers:
transl_table = int(feature.qualifiers['transl_table'][0])
p = mRNA.seq.translate(table=transl_table, cds=True)
except:
print("Protein {0} in gene {1} could not be translated!".
format(feature.qualifiers['protein_id'][0], seqrec.id))
print(feature.qualifiers[438369.1])
print(n_cds)
</code>
|
{
"filename": "biopython_notebook_1.ipynb",
"repository": "Deya-B/Bioinformatics-notes",
"query": "transformed_from_existing",
"size": 199361,
"sha": ""
}
|
# 02-warmup-sol.ipynb
Repository: hanisaf/mist5730-6380-spring2020
Refer to [the University of Georgia by the Numbers Page](https://www.uga.edu/facts.php)
Reconstruct (most) of this page using markdown in this notebook
# UGA by the Numbers
**Founded:**
> January 27, 1785, by the Georgia General Assembly. UGA is the birthplace of public higher education in America.
**Location:**
> Athens, Georgia (Clarke County), about 60 miles northeast of downtown Atlanta.
**Size:**
> Main campus: 465 buildings on 762 acres.
> Total acreage in 31 Georgia counties: 39,743 acres
**Work Force**
> | | |
> |----------------------------------------------------------|-------------------|
> | **Faculty (instruction/research/public service)** | 3,119 |
> | **Administrative/other professional** | 3,213 |
> | **Technical/clerical/crafts/maintenance** | 4,524 |
> | **Total** | **10,856** |
**Annual Budget (FY 2018):**
> $1.64 billion
**Research (FY 2017)**
> Research and development expenditures: \$455 million
>
> Licensing Revenue: $10.6 million
**Schools and Colleges**
> The University of Georgia is a comprehensive land and sea grant institution composed of 17 schools and colleges. They are, in order of founding:
>
> - Franklin College of Arts and Sciences, 1801
> - College of Agricultural and Environmental Sciences, 1859
> - School of Law, 1859
> - College of Pharmacy, 1903
> - Daniel B. Warnell School of Forestry and Natural Resources, 1906
> - College of Education, 1908
> - Graduate School, 1910
> - C. Herman and Mary Virginia Terry College of Business, 1912
> - Henry W. Grady College of Journalism and Mass Communication, 1915
> - College of Family and Consumer Sciences, 1918
> - College of Veterinary Medicine, 1946
> - School of Social Work, 1964
> - College of Environment and Design, 1969
> - School of Public and International Affairs, 2001
> - College of Public Health, 2005
> - Eugene P. Odum School of Ecology, 2007
> - College of Engineering, 2012
>
> The university is also home to the Augusta University/University of Georgia Medical Partnership.
|
{
"filename": "02-warmup-sol.ipynb",
"repository": "hanisaf/mist5730-6380-spring2020",
"query": "transformed_from_existing",
"size": 3939,
"sha": ""
}
|
# SIMS_tutorial_4.ipynb
Repository: braingeneers/SIMS
## **SIMS Tutorial**
In this tutorial, we will walk through the [SIMS (Scalable, Interpretable Machine Learning for Single Cell)](https://www.cell.com/cell-genomics/fulltext/S2666-979X(24)00165-4) pipeline step by step. SIMS is a deep learning-based tool built on TabNet, a transformer-based model optimized for tabular data. It is designed to classify single-cell RNA sequencing (scRNA-seq) data while providing interpretable predictions.
By following this tutorial, you will learn how to:
- Download and prepare scRNA-seq data for SIMS
- Train a SIMS model to classify cell types
- Use the trained model to make predictions on new, unseen data
- Evaluate predictions using accuracy metrics and visualization tools
### **Before you begin**
The `scsims` package is only compatible with Python versions between 3.8 and 3.11. Run the following cell to ensure you are using a compatible version (we recommend Python 3.9 for optimal performance).
<code>
!python --version
</code>
If you're running this notebook for the first time, uncomment and execute the following line to **install the required packages:**
<code>
# !pip install --use-pep517 git+https://github.com/braingeneers/SIMS.git
</code>
Once the packages are installed, we can **import them:**
<code>
import scanpy as sc
import pandas as pd
import anndata as ad
import os
import torch
from scsims import SIMS
from pytorch_lightning.callbacks import EarlyStopping, ModelCheckpoint
from sklearn.model_selection import train_test_split
</code>
### **Step 1: Prepare data**
Before we can train our model, we need to prepare the data. This involves several key steps to ensure that the dataset is structured correctly and optimized for machine learning.
#### `1.a` Download data
The dataset we are using comes from a study published in eLife by Domingo-Gonzalez et al. (2020). This study investigated immune cells in the developing mouse lung at single-cell resolution. It contains only 4,052 cells, making it ideal for this notebook (large enough for meaningful training but small enough for computational efficiency).
🔗 Source:
- [UCSC Cell Browser website](https://cells.ucsc.edu/?bp=lung&org=Mouse+(M.+musculus)) – where we download the dataset.
- [Domingo-Gonzalez et al., 2020 (eLife)](https://elifesciences.org/articles/56890) – original study providing the data.
<code>
!curl -O https://cells.ucsc.edu/mouse-lung-immune/exprMatrix.tsv.gz
!curl -O https://cells.ucsc.edu/mouse-lung-immune/meta.tsv
</code>
#### `1.b` Load the Anndata object
In this step, we will load the gene expression matrix and associated metadata, clean the data, and convert it into an AnnData object for further processing.
<code>
# Load expression matrix
expr_matrix = pd.read_csv("exprMatrix.tsv.gz", sep="\t", index_col=0)
# Load metadata
meta = pd.read_csv("meta.tsv", sep="\t", index_col=0)
# This particular dataset stores gene names in a 'GENE|GENE' format
# This step removes duplicate gene names if they exist
if "|" in expr_matrix.index[0]:
expr_matrix.index = expr_matrix.index.str.split('|').str[0]
# Transpose to make cells as rows and genes as columns (CELL x GENE)
expr_matrix = expr_matrix.T
# Ensure indices match
expr_matrix = expr_matrix.loc[meta.index]
# Create AnnData object
adata = ad.AnnData(expr_matrix)
# Add metadata to AnnData object
adata.obs = meta
# Show basic info
print(adata)
</code>
#### `1.c` Select target feature
In this example dataset, cell type labels are stored in the 'Cell Subtype' column, so we will remove other columns as they are not needed for training.
🔹 **Note:** If you're using a different dataset, replace 'Cell Subtype' with the appropriate column that contains your cell type annotations. Run `adata.obs.columns` to check available columns in your dataset before selecting the target feature.
<code>
class_label = 'Cell Subtype'
adata.obs = adata.obs[[class_label]] # Filter out other columns
unique_classes = adata.obs[class_label].unique()
print("Cell types: ", unique_classes)
</code>
#### `1.d` Preprocess the data
Now we need to preprocess the data to ensure it is clean, normalized, and ready for model training.
🔹 **Important:** The same preprocessing steps must be applied consistently to both the training dataset and any new data used for inference to ensure compatibility.
<code>
# Perform some light filtering
sc.pp.filter_cells(adata, min_genes=100)
sc.pp.filter_genes(adata, min_cells=3)
# Transform the data for model ingestion
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.scale(adata)
</code>
#### `1.e` Split data
In real-world applications, SIMS is typically used with two datasets: a **labeled** dataset for training and an **unlabeled** dataset for generating predictions. Since we don't have a separate unlabeled dataset for this example, we'll split our labeled dataset into a **training set** (used to train the model) and a **test set** (used to evaluate its performance). This allows us to assess how well SIMS generalizes to unseen data.
🔹 **Note:** If you are using your own dataset, do not split it. Instead, load your test data separately during the prediction step. Make sure to preprocess your test data using the same steps applied to your training data to maintain consistency and avoid errors.
<code>
# Split cells into train and test (ex: 80% train, 20% test)
train_cells, test_cells = train_test_split(adata.obs_names, test_size=0.2, random_state=42)
# Create training and "unlabeled" test datasets
adata_train = adata[train_cells].copy()
adata_test = adata[test_cells].copy()
</code>
### **Step 2: Train the model with SIMS**
Now that our dataset is preprocessed, we can train a machine learning model using SIMS. SIMS is built on [TabNet](https://arxiv.org/abs/1908.07442), a deep learning architecture optimized for tabular data. It allows us to classify immune cells based on gene expression while maintaining model interpretability.
#### `2.a` Initialize SIMS model
We first create a SIMS object using the training dataset (`adata_train`) and specify the cell type label column:
<code>
sims = SIMS(data=adata_train, class_label=class_label)
</code>
#### `2.b` Set up model parameters
Next, we configure the model using setup_model():
<code>
sims.setup_model(n_a=64, n_d=64, weights=sims.weights)
</code>
- `n_a=64` and `n_d=64` define the number of attention and decision steps in the TabNet architecture.
- `weights=sims.weights` ensures that the model adjusts for imbalanced cell types by weighting the loss inversely to label frequency. This helps the model learn rare cell types more effectively.
#### `2.c` Define Checkpointing Strategy
Since training deep learning models takes time, we save the best version using a checkpointing system:
<code>
checkpoint_callback = ModelCheckpoint(
dirpath="./sims", # Save in the notebook's current directory
filename="sims_model", # File will be saved as 'sims_model.ckpt'
save_top_k=1, # Keep only the best checkpoint
monitor="val_loss", # Save the best model based on validation loss
mode="min", # Lower validation loss is better
)
</code>
#### `2.d` Configure the Training Process
We set up the training loop using a PyTorch Lightning trainer:
<code>
sims.setup_trainer(
callbacks=[
EarlyStopping(monitor="val_loss", patience=50), # Stop training if validation loss does not improve for 50 epochs
checkpoint_callback, # Save the best model
],
max_epochs=50, # Complete training cycles through the dataset, can increase
accelerator="cpu", # Forces training on CPU, can switch to GPU/MPS if available
devices=1, # Uses one CPU/GPU device
logger=False, # Disable/enable logging
)
</code>
🔹 **Note:** Weights & Biases (WandB) logging has been disabled in this example for simplicity. However, if you want to use WandB logging, follow these steps **before configuring the training process** (in the previous cell):
1. Import WandbLogger: `from pytorch_lightning.loggers import WandbLogger`
2. Initialize and set offline to True: `logger = WandbLogger(offline=True)`
3. In `sims.setup_trainer()`, change code below to `logger=logger`
#### `2.e` Train and save model
Once everything is set up, we train the model with:
<code>
sims.train()
</code>
After training, we verify that a trained model was saved:
<code>
print("Saved model:", [f for f in os.listdir() if f.endswith(".ckpt")])
</code>
### **Step 3: Predict on new data with trained model**
#### `3.a` Load the Trained Model
To make predictions, we first load the trained SIMS model from the checkpoint:
<code>
sims = SIMS(weights_path="sims_model.ckpt", map_location=torch.device('cpu'))
</code>
- `weights_path="sims_model.ckpt"` loads the model checkpoint.
- `map_location=torch.device('cpu')` ensures the model runs on CPU (set to "cuda" if using a GPU).
#### `3.b` Predict cell types
We then use the model to predict cell types for the test dataset (`adata_test`). This returns a DataFrame containing predicted labels for each cell.
<code>
cell_predictions = sims.predict(adata_test)
</code>
#### `3.c` Align predictions with cell names, add predictions to AnnData object, and view
To ensure that the predictions are correctly indexed, we align them with `adata_test.obs_names`. We then merge the predictions with the metadata of the test dataset (`adata_test.obs`) and can view the predictions.
<code>
cell_predictions.index = adata_test.obs_names # Align predictions with `adata_test`
adata_test.obs = adata_test.obs.join(cell_predictions, rsuffix='_pred')
print(adata_test.obs.head())
</code>
### **Step 4: Evaluate and visualize**
Once we have predicted cell types, we can evaluate how well the model performed. Each of these steps is optional.
#### `4.a` Calculate model accuracy
We compute accuracy, which measures the proportion of correctly classified cells:
<code>
from sklearn.metrics import accuracy_score
accuracy = accuracy_score(adata_test.obs[class_label], adata_test.obs["pred_0"])
print(f"Model Accuracy: {accuracy:.2f}")
</code>
#### `4.b` Generate a classification report
A classification report provides detailed performance metrics for each cell type, such as:
- Precision – How many of the predicted cells are actually correct?
- Recall – How many of the true cells were correctly classified?
- F1-score – A balanced metric combining precision and recall.
<code>
from sklearn.metrics import classification_report
report = classification_report(
adata_test.obs[class_label],
adata_test.obs["pred_0"],
zero_division=0 # Set to 0 instead of raising a warning
)
print(report)
</code>
#### `4.c` Compute and visualize confusion matrix
To see how predictions compare to actual labels, we compute a confusion matrix, where:
- Each row represents actual cell types.
- Each column represents predicted cell types.
- Diagonal values indicate correct classifications, while off-diagonal values show misclassifications.
<code>
from sklearn.metrics import confusion_matrix
import matplotlib.pyplot as plt
import seaborn as sns
# Compute confusion matrix
cm = confusion_matrix(adata_test.obs[class_label], adata_test.obs["pred_0"])
# Normalize by row (convert counts to percentages)
cm_normalized = cm.astype("float") / cm.sum(axis=1, keepdims=True)
# Get all unique cell types
labels = sorted(adata_test.obs[class_label].unique())
# Plot normalized confusion matrix
plt.figure(figsize=(8, 6))
sns.heatmap(cm_normalized, annot=True, fmt=".2f", xticklabels=labels, yticklabels=labels, cmap="Blues")
# Add labels and title
plt.xlabel("Predicted")
plt.ylabel("True")
plt.title("Normalized Confusion Matrix (Row Percentage)")
plt.show()
</code>
### **Step 5: View Explainability**
Interpreting machine learning models is critical, especially in biological research, where understanding why a model makes predictions can reveal important biological insights. SIMS uses TabNet's built-in explainability to identify which genes contribute most to its predictions.
The explainability matrix assigns an importance score to each gene based on how much it influenced the model's classification decisions. This allows us to determine the top contributing genes and assess whether they align with known biological markers.
#### `5.a` Generate the Explainability Matrix
We will use `sims.explain()` to compute feature importance scores for each gene. This produces an explainability matrix, where each value represents how much a specific gene contributed to the model’s classification decision for a given cell. We then convert it to a pandas DataFrame for easy manipulation and compute the average importance score for each gene across all cells.
<code>
# Run explainability analysis
explainability_matrix = sims.explain(adata_test, num_workers=0, batch_size=32)[0]
# Convert explainability matrix to a DataFrame
explain_df = pd.DataFrame(explainability_matrix, columns=sims.model.genes)
# Compute average gene importance
gene_importance = explain_df.mean(axis=0)
</code>
#### `5.b` View most influential genes
Now that we have computed gene importance scores, we can identify and print the top n most influential genes (the ones that contributed the most to the model’s classification decisions).
<code>
top10_genes = gene_importance.nlargest(10) # Can increase to top 20, 30, etc.
print("Top 10 most important genes:")
print(top10_genes)
</code>
|
{
"filename": "SIMS_tutorial_4.ipynb",
"repository": "braingeneers/SIMS",
"query": "transformed_from_existing",
"size": 119177,
"sha": ""
}
|
# HiDENSEC.ipynb
Repository: songlab-cal/HiDENSEC
# Global Variables & Function Definitions
These global definitions require evaluation before running HiDENSEC on any concrete Hi-C map.
## Modules
<code>
import numpy as np
import scipy.sparse as sp_sparse
import scipy.signal as sp_signal
import scipy.ndimage as sp_image
from scipy.stats import kendalltau
import scipy.stats as sp_stats
</code>
<code>
rng = np.random.default_rng()
</code>
## Global variables
Load file paths, covariates & centromere locations.
<code>
chromosomelocations = np.loadtxt('chromosomelocations.txt')-1
compartmentnames = np.loadtxt('compartmentnames.txt', dtype='str')
compartments = np.loadtxt('compartments.txt', dtype='str')
centromers = np.loadtxt('centromers.txt')-1
newCentromers = np.loadtxt('newCentromers.txt')-1
rPos = np.loadtxt('rPos.txt')-1
fPos = np.loadtxt('fPos.txt')-1
acrox = np.loadtxt('acrox.txt')
corrchromlocations = np.loadtxt('corrchromlocations.txt')-1
centromersCorrected = np.loadtxt('centromersCorrected.txt')-1
covariates = np.loadtxt('covariates.csv', delimiter=',')
excursionLengthsH0 = np.loadtxt('excursionLengthsH0.csv', delimiter=',')
fixCdata = np.loadtxt('fixCdata.csv', delimiter=',', dtype='object')
hiCdata = np.loadtxt('hiCdata.csv', delimiter=',', dtype='object')
</code>
## Covariate Correction
Function definitions pertaining to covariate regression.
<code>
def partitionByIndex(list, indices):
return np.split(list, indices)
def partitionByLength(list, lengths):
return partitionByIndex(list, np.cumsum(lengths))
def findDiploidPeak(data, window=[0.1,1,0.1]):
clean_data = data[data>0]
scale = np.median(np.abs(clean_data - np.median(clean_data)))
modes = []
for c in np.arange(window[0], window[1] + window[2], window[2]):
counts, bins = np.histogram(clean_data, bins=np.arange(clean_data.min(), clean_data.max(), c*scale))
mode_index = int(np.median(np.argmax(counts)))
modes.append((bins[mode_index] + bins[mode_index+1])/2)
return np.mean(modes)
def filterPosition(dataset, cutthreshold=75, GCthreshold=0.32, mapthreshold=0.8):
covariate_part = dataset[:, 1:4].astype(float)
compartment_part = dataset[:,4]
return (covariate_part[:,0]>cutthreshold) & (covariate_part[:,1]>GCthreshold) & (covariate_part[:,2]>mapthreshold) & (compartment_part!='A0')
def covariateFilter(dataset, cutthreshold=75, GCthreshold=0.32, mapthreshold=0.8):
return dataset[filterPosition(dataset, cutthreshold, GCthreshold, mapthreshold)]
def covariateCorrection1(dataset, cutthreshold=75, GCthreshold=0.32, mapthreshold=0.8, midpointwindow=[0.1,1,0.1], neighbourhood=0.3, filterlength=100):
data = covariateFilter(dataset, cutthreshold, GCthreshold, mapthreshold)
midpoints = [ findDiploidPeak(data[data[:,4] == compartment_name, 0].astype(float), midpointwindow) for compartment_name in compartmentnames[1:] ]
inserted_midpoints = np.copy(data[:,-1])
for j in range(5):
inserted_midpoints[inserted_midpoints == compartmentnames[1+j]] = midpoints[j]
compartmentcorrected = 2*data[:,0].astype(float)/inserted_midpoints.astype(float)
predictions = np.copy(data[:, -1])
for compartment_name in compartmentnames[1:]:
compartment_indices = (data[:, -1] == compartment_name) & (np.abs(compartmentcorrected - 2) < neighbourhood)
compartment_covariates = data[compartment_indices][:, [1,2]].astype(float)
compartment_values = compartmentcorrected[compartment_indices].astype(float)
design_mat = np.concatenate( [np.ones([compartment_indices.sum(),1]), compartment_covariates], axis=1 )
fit = np.linalg.lstsq(design_mat, compartment_values, rcond=None)
full_compartment_indices = (data[:, -1] == compartment_name)
full_compartment_covariates = data[full_compartment_indices][:, [1,2]].astype(float)
full_design_mat = np.concatenate( [np.ones([full_compartment_indices.sum(),1]), full_compartment_covariates], axis=1 )
predictions[predictions == compartment_name] = full_design_mat @ fit[0]
fully_corrected_data = 2*compartmentcorrected / predictions.astype(float)
fully_corrected_data = 2*fully_corrected_data / findDiploidPeak(fully_corrected_data)
return fully_corrected_data
def attachCovariates(profile):
return np.concatenate([profile[:,None], np.transpose(covariates), compartments[:,None]], axis=1)
def rawToCorrected(x, filter):
if type(x) == int:
return int(np.median(np.argmin(np.abs(x-filter))))
else:
return [int(np.median(np.argmin(np.abs(y-filter)))) for y in x]
def xToChi(x):
return np.argmax(x < corrchromlocations[1:])+1
def dataCcorrector(profile, data_flag, cutthreshold=75, GCthreshold=0.32, mapthreshold=0.8, midpointwindow=[0.1,1,0.1], neighbourhood=0.3, filterlength=100):
test_data = attachCovariates(profile)
test_data = covariateFilter(test_data, cutthreshold, GCthreshold, mapthreshold)
if data_flag == 'HiC':
reference_data = np.copy(hiCdata)
elif data_flag == 'FixC':
reference_data = np.copy(fixCdata)
else:
return 'Unknown protocol'
reference_data = reference_data[:, [4, 1, 2, 3, 0]]
data = covariateFilter(reference_data, cutthreshold, GCthreshold, mapthreshold)
midpoints = [ findDiploidPeak(data[data[:,4] == ('"'+compartment_name+'"'), 0].astype(float), midpointwindow) for compartment_name in compartmentnames[1:] ]
inserted_midpoints = np.copy(data[:,-1])
for j in range(5):
inserted_midpoints[inserted_midpoints == ('"'+compartmentnames[1+j]+'"')] = midpoints[j]
compartmentcorrected = 2*data[:,0].astype(float)/inserted_midpoints.astype(float)
test_inserted_midpoints = np.copy(test_data[:,-1])
for j in range(5):
test_inserted_midpoints[test_inserted_midpoints == (compartmentnames[1+j])] = midpoints[j]
test_compartmentcorrected = 2*test_data[:,0].astype(float)/test_inserted_midpoints.astype(float)
predictions = np.copy(test_data[:, -1])
for compartment_name in compartmentnames[1:]:
compartment_indices = (data[:, -1] == ('"'+compartment_name+'"')) & (np.abs(compartmentcorrected - 2) < neighbourhood)
compartment_covariates = data[compartment_indices][:, [1,2]].astype(float)
compartment_values = compartmentcorrected[compartment_indices].astype(float)
design_mat = np.concatenate( [np.ones([compartment_indices.sum(),1]), compartment_covariates], axis=1 )
fit = np.linalg.lstsq(design_mat, compartment_values, rcond=None)
full_compartment_indices = (test_data[:, -1] == compartment_name)
full_compartment_covariates = test_data[full_compartment_indices][:, [1,2]].astype(float)
full_design_mat = np.concatenate( [np.ones([full_compartment_indices.sum(),1]), full_compartment_covariates], axis=1 )
predictions[predictions == compartment_name] = full_design_mat @ fit[0]
fully_corrected_data = 2*test_compartmentcorrected / predictions.astype(float)
fully_corrected_data = 2*medianFilter(fully_corrected_data, 100) / findDiploidPeak(medianFilter(fully_corrected_data, 100))
return fully_corrected_data
</code>
## Copy number & Mixture Proportion Estimation
Function definitions calculating effective copy number profiles.
<code>
def medianDeviation(list):
return np.median(np.abs(list - np.median(list)))
def splitList(list, index):
return ([list[:index], list[index:]], [index / len(list), 1-index/len(list)])
def findRestrictedMedian(list, candidates):
epsilon_list = [np.mean(np.abs(list - candidate)) for candidate in candidates]
minPos = np.argmin(epsilon_list)
return (candidates[minPos], epsilon_list[minPos], medianDeviation(list))
def findSplit(data, candidates):
restricted_medians = []
for j in range(2, len(data) - 1):
splits = splitList(data, j)
split_deviations = [np.sum(findRestrictedMedian(split, candidates)[1:]) for split in splits[0]]
restricted_medians.append(np.array(split_deviations)@np.array(splits[1]))
return (np.argmin(restricted_medians)+1, np.min(restricted_medians))
def paddedPartition(list, window):
a = [list[:window] for _ in range(round((window-1)/2))]
b = [list[j:window+j] for j in range(len(list)-window+1)]
c = [list[:window] for _ in range(round((window-1)/2))]
return np.concatenate([a,b,c])
def copyNumberFilter(data, candidates, window=101):
return [findRestrictedMedian(part, candidates) for part in paddedPartition(data, window)]
def copyNumberVariance(data, candidates, window=101):
restricted_medians = [findRestrictedMedian(part, candidates) for part in paddedPartition(data, window)]
return np.mean([z[0]*z[1]/z[2] for z in restricted_medians])
def listCandidates(f, maxPloidy):
return 2*(1-f)+f*np.arange(1, maxPloidy+1)
def computeChangePoints(profile):
return np.arange(len(profile)-1)[np.abs(np.diff(profile)) > 0]
def refineChangePoint(data, pt, candidates, window=250, replicates=100):
w = round((window-1)/2)
split = findSplit(data[pt-w:pt+w+1], candidates)
a = pt-w-1+split[0]
replicates = [findSplit(rng.choice(data[pt-w:pt+w+1], 2*w+1), candidates)[-1] for _ in range(replicates)]
return (a, (replicates > split[-1]).sum() / len(replicates))
def refineChangePoints(data, pts, candidates, window=250, replicates=100):
extended_pts = np.concatenate(([0], pts, [len(rPos)-1]))
parts = [extended_pts[j:j+3] for j in range(len(extended_pts)-2)]
ws = [ np.min(np.append(np.diff(part)/2, window)) for part in parts ]
res = []
for pt, w in zip(pts, ws):
if w <= 3.5:
res.append((pt, 0))
else:
res.append(refineChangePoint(data, pt, candidates, w, replicates))
return res
def refineProfile(profile, pts):
extended_pts = np.concatenate(([0], pts, [len(rPos)]))
refined_segments = []
for j in range(len(extended_pts)-1):
a = int(extended_pts[j])
b = int(extended_pts[j+1])
values, counts = np.unique(profile[a:b+1], return_counts=True)
index = np.argmax(counts)
refined_segments = np.append(refined_segments, [values[index] for _ in range(b-a)])
return refined_segments
def flattenExcursions(profile, threshold=200):
pts = computeChangePoints(profile)
partitioned_profile = partitionByIndex(profile, pts)
pos = np.array([ (len(part) <= threshold) for part in partitioned_profile])
flattened_profile = []
for j, part in enumerate(partitioned_profile):
n = len(part)
if (n<=threshold) & (0 < j < len(partitioned_profile)-1):
flattened_profile.append(np.concatenate(([partitioned_profile[j-1][0] for _ in range(round(n/2))], [partitioned_profile[j+1][0] for _ in range(round((n+1)/2)) ])))
else:
flattened_profile.append(partitioned_profile[j])
return np.concatenate(flattened_profile)
def rankExcursions(data, refinedProfile, changePoints):
partitioned_profile = partitionByIndex(refinedProfile, changePoints[:,0].astype(int))
partitioned_data = partitionByIndex(data, changePoints[:,0].astype(int))
H0 = data[(1.99 <= refinedProfile) & (refinedProfile <= 2.01)]
extended_change_points = np.concatenate([[0.5], changePoints[:,1], [0.5]])
intervalP = [(extended_change_points[j] + extended_change_points[j+1]) for j in range(len(extended_change_points)-1)]
def extractBulk(list):
n = np.max([round(0.2*len(list)),1])
return list[n:-n]
def findClosestDiploidBlock(blocks, k):
a, b = np.nonzero(blocks < k)[0], np.nonzero(blocks > k)[0]
if len(a)>0:
if len(b)>0:
return [blocks[a[-1]], blocks[b[0]]]
else:
return [blocks[a[-1]]]
elif len(b)>0:
return [blocks[b[0]]]
else:
return []
block_ploidy = np.array([part[0] for part in partitioned_profile])
diploidBlocks = np.nonzero((block_ploidy <= 2.01) & (1.99 <= block_ploidy))[0]
stat_vals_1 = []
for part_profile, part_data, j in zip(partitioned_profile, partitioned_data, range(len(partitioned_profile))):
a = np.mean((excursionLengthsH0 <= len(part_profile)))
sample_data = np.concatenate([partitioned_data[j] for j in findClosestDiploidBlock(diploidBlocks, j)])
sampled_data = rng.choice(sample_data, size=(100, len(extractBulk(part_profile))))
dataH0 = [medianDeviation(sampled_point) for sampled_point in sampled_data]
threshold = 2 / part_profile[0] * medianDeviation(extractBulk(part_data))
b = np.mean(dataH0 > threshold)
stat_vals_1.append(np.max([a,b])**2)
def uniformSumCDF(x):
return np.piecewise(x, [x < 0, (0 <= x) & (x < 1), (1 <= x) & (x < 2), 2 <= x], [0, lambda x: x**2/2, lambda x: 1-0.5*(2-x)**2, 1])
stat_vals_2 = uniformSumCDF(np.array([interval.sum() for interval in intervalP]))
stat_vals = stat_vals_1 + stat_vals_2
return uniformSumCDF(stat_vals)
def medianFilter(data, window):
return np.array([np.median(data[j:j+window]) for j in range(len(data)-window+1)])
def extractExcursions(profile, points):
extended_points = np.concatenate([[0], points, [len(rPos)]])
excursion_indices = [[extended_points[j], extended_points[j+1]] for j in range(len(extended_points)-1)]
excursions = partitionByIndex(profile, points)
excursion_means = np.array([np.mean(excursion) for excursion in excursions])
return np.arange(len(excursion_indices))[np.abs(excursion_means - 2) > 0.001]
def excursionLengths(profile, points):
pos = extractExcursions(profile, points)
return [len(partitionByIndex(profile, points)[j]) for j in pos]
def benjaminiHochberg(pvalues, alpha):
ps = np.sort(1-pvalues) - alpha / len(pvalues) * np.arange(1, len(pvalues)+1)
indices = np.nonzero(ps > 0)[0]
if len(indices) == 0:
return 0
else:
return 1+indices[0]
def confidenceInterval(data, profile, f, resample_size=100):
pts = computeChangePoints(profile)
partitioned_profile = partitionByIndex(profile, pts)
partitioned_data = partitionByIndex(data, pts)
pos = extractExcursions(profile, pts)
partitioned_profile_pos = [partitioned_profile[j] for j in pos]
excursion_ploidy = np.abs([ 2 - (np.unique(profile_part)[0] - 2*(1-f)) / 2 for profile_part in partitioned_profile_pos ])
excursion_data = [partitioned_data[j] for j in pos]
excursion_lengths = np.array([len(profile_part) for profile_part in partitioned_profile_pos])
resample_sizes = resample_size * excursion_lengths / np.sum(excursion_lengths)
fluctuations = []
for data_point, size, ploidy in zip(excursion_data, resample_sizes, excursion_ploidy):
fluctuations = np.append(fluctuations, [np.abs(np.median(rng.choice(data_point, len(data_point))) - 2) / ploidy for _ in range(int(size))])
return 2*medianDeviation(fluctuations)
def findThreshold(pvalues, alpha):
delta = 1
iter = 1
sorted_ps = np.sort(pvalues)[::-1]
while (delta>0) & (iter<np.min([1000, len(pvalues)-1])):
delta = benjaminiHochberg(sorted_ps[:iter+1], alpha) - benjaminiHochberg(sorted_ps[:iter], alpha)
iter+=1
return iter
def estimate_proportion_ploidy(rowsums, maxPloidy):
data = rowsums
y = int(corrchromlocations[10])
smoothed_data = 2*medianFilter(data[:y], 100) / findDiploidPeak(medianFilter(data[:y], 100))
sigma = []
for j in tqdm(np.arange(0,1.01,0.01)):
sigma.append(copyNumberVariance(smoothed_data, 2*(1-j) + j*np.arange(1, maxPloidy+1), 251))
f = np.arange(0, 1.01, 0.01)[np.argmin(sigma)]
pi0 = np.array(copyNumberFilter(rowsums, 2*(1-f) + f*np.arange(1, maxPloidy+1), 501))
xs = np.array(refineChangePoints(data, computeChangePoints(pi0[:,0]), listCandidates(f, maxPloidy), 250))
pi1 = refineProfile(pi0[:,0], xs[:,0])
ps = rankExcursions(data, pi1, xs)
return pi1, f, ps
</code>
## Off-diagonal detection
Function definitions detecting fusion events of type (a) and (b)
<code>
def roundToChromosome(x,y):
chiRange = [xToChi(z) for z in range(x,y+1)]
chis, counts = np.unique(chiRange, return_counts=True)
chi = int(np.median(chiRange))
if np.max(counts / np.sum(counts)) < 0.6:
acs = []
for j in chis:
a = np.max([x, corrchromlocations[j-1]])
b = np.min([y, corrchromlocations[j] - 1])
if corrchromlocations[xToChi(b)]-1-b < 50:
c = corrchromlocations[xToChi(b)] - 1
else:
c = b
acs.append([a,c])
acs = np.array(acs, dtype=int)
acs = acs[(acs[:,1] - acs[:,0]) > 200]
return acs
else:
chi_nearest_range = np.arange(corrchromlocations[chi-1], corrchromlocations[chi]-1)
nearest_x = chi_nearest_range[np.abs(chi_nearest_range - x).argmin()]
nearest_y = chi_nearest_range[np.abs(chi_nearest_range - y).argmin()]
if corrchromlocations[xToChi(nearest_x)]-1-nearest_x < 50:
a = corrchromlocations[xToChi(nearest_x)]-1
else:
a = nearest_x
if corrchromlocations[xToChi(nearest_y)]-1-nearest_x < 50:
b = corrchromlocations[xToChi(nearest_y)]-1
else:
b = nearest_y
return np.array([a, b], dtype=int)
def scanBlock1(block, ws):
ws_prod = np.product(ws)
return sp_signal.oaconvolve(np.ones(ws), block.toarray(), mode='valid') / ws_prod
def scanBlock2(mat, chi1, chi2, ws):
a = corrchromlocations[chi1-1]
b = corrchromlocations[chi1]
c = corrchromlocations[chi2-1]
d = corrchromlocations[chi2]
ws_prod = np.product(ws)
return sp_signal.oaconvolve(np.ones(ws), mat.toarray()[int(a):int(b), int(c):int(d)], mode='valid') / ws_prod
def computeIntensitySquare(mat, x, y, ws):
a = np.max([x-ws[0]+1, 0])
b = np.min([x+ws[0]+1, len(rPos)])
c = np.max([y-ws[1]+1, 0])
d = np.min([y+ws[1]+1, len(rPos)])
mat11 = mat[a:x+1, c:y+1]
mat12 = mat[a:x+1, y+1:d]
mat21 = mat[x+1:b, c:y+1]
mat22 = mat[x+1:b, y+1:d]
return np.array([[mat11.mean(), mat12.mean()], [mat21.mean(), mat22.mean()]])
def detectPattern(intensitySquare):
normalization_constant = intensitySquare.sum()
if normalization_constant <= 10**(-8):
return 0.
else:
normalized_densities = np.concatenate(intensitySquare) / normalization_constant
return np.max(normalized_densities)
def rowPattern(mat, x, ws):
chi = xToChi(x)
n = len(rPos)
w = ws[-1]
y_indices = np.concatenate([np.arange(0, corrchromlocations[chi-1]), np.arange(corrchromlocations[chi], n)])
r_block = mat[x-ws[0]+1:x+1, y_indices]
r = np.concatenate(scanBlock1(r_block, ws))
s_block = mat[x+1:x+ws[0]+1, y_indices]
s = np.concatenate(scanBlock1(s_block, ws))
detected_patterns = []
for j in range(len(r)-w):
pattern_mat = np.array([[r[j], r[j+w]], [s[j], s[j+w]]])
detected_patterns.append(detectPattern(pattern_mat))
return np.array(detected_patterns)
def testTreeStructure(mat, x, y, w, resamples=100):
submatrix = mat[np.max([x-w[0],0]):np.min([x+w[0]+1, len(rPos)]), np.max([y-w[1],0]):np.min([y+w[1]+1, len(rPos)])]
rowMarginals = submatrix.mean(axis=1)
columnMarginals = submatrix.mean(axis=0)
rowStatistic = np.mean([np.var(rowMarginals[:w[0]]) * w[0] / (w[0]-1), np.var(rowMarginals[w[0]+1:]) * len(rowMarginals[w[0]+1:]) / (len(rowMarginals[w[0]+1:]) - 1)])
columnStatistic = np.mean([np.var(columnMarginals[:w[0]]) * w[0] / (w[0]-1), np.var(columnMarginals[w[0]+1:]) * len(columnMarginals[w[0]+1:]) / (len(columnMarginals[w[0]+1:]) - 1)])
resampledRowStatistic = []
resampledColumnStatistic = []
for _ in range(resamples):
permutedRows = rng.permutation(rowMarginals)
permutedColumns = rng.permutation(columnMarginals)
resampledRowStatistic.append(np.mean([np.var(permutedRows[:w[0]]) * w[0] / (w[0]-1),
np.var(permutedRows[w[0]+1:]) * len(permutedRows[w[0]+1:]) / (len(permutedRows[w[0]+1:]) - 1)]))
resampledColumnStatistic.append(np.mean([np.var(permutedColumns[:w[0]]) * w[0] / (w[0]-1),
np.var(permutedColumns[w[0]+1:]) * len(permutedColumns[w[0]+1:]) / (len(permutedColumns[w[0]+1:]) - 1)]))
return np.mean([np.mean(resampledRowStatistic > rowStatistic), np.mean(resampledColumnStatistic > columnStatistic)])
def extractButterflyCandidate(mat, chi1, chi2, ws, componentDepth=10):
blockScan = scanBlock2(mat, chi1, chi2, ws)
components = sp_image.label(np.where(blockScan > np.median(blockScan), blockScan, 0))[0]
component_groups, component_counts = np.unique(components, return_counts=True)
size_threshold = np.sort(component_counts)[-componentDepth]
large_groups = component_groups[component_counts >= size_threshold]
large_group_sizes = component_counts[component_counts >= size_threshold]
group_maxs = [[np.unravel_index(np.where(components == group, blockScan, 0).argmax(), blockScan.shape), round(np.sqrt(size/2))]
for group, size in zip(large_groups, large_group_sizes)]
return group_maxs
def pickLargeCandidate(mat, x, y, ws):
localMat = mat[np.max([x-ws[0], 1]):np.min([x+ws[0], len(rPos)]), np.max([y-ws[1], 1]):np.min([y+ws[1], len(rPos)])]
return np.array(np.unravel_index(localMat.argmax(), localMat.shape)) + np.array([1+x,1+y]) - np.array([1+ws[0], 1+ws[1]])
def butterflySummary(mat, x, y, ws):
intensitySquare = computeIntensitySquare(mat, x, y, ws)
if intensitySquare.sum() <= 0:
intensitySquare = np.zeros([2,2])
else:
intensitySquare = intensitySquare / intensitySquare.sum()
return -np.linalg.det(intensitySquare)
def findButterflySummary(mat, x, y, ws, symmetry=0):
a = np.max([0, x-ws[0]])
b = np.min([len(rPos), x+ws[0]+1])
c = np.max([0, y-ws[1]])
d = np.min([len(rPos), y+ws[1]+1])
localMat = np.array([[butterflySummary(mat, j, k, [50, 50]) for k in np.arange(c,d)] for j in np.arange(a,b)])
if symmetry == 0:
argmax = np.unravel_index(localMat.argmax(), localMat.shape)
return [np.array(argmax) + np.array([x, y]) - np.array(ws), localMat[argmax]]
else:
argmin = np.unravel_index(localMat.argmin(), localMat.shape)
return [np.array(argmin) + np.array([x, y]) - np.array(ws), localMat[argmin]]
return localMat
def treeStatistic(mat, x, y, w):
a11 = mat[np.max([0, x-w-1]):x, np.max([0, y-w]):y+1][::-1]
a12 = np.transpose(mat[np.max([0, x-w-1]):x, y+1:np.min([len(rPos), y+w+2])])
a21 = mat[x:np.min([len(rPos), x+w+1]), np.max([0, y-w]):y+1]
a22 = np.transpose(mat[x:np.min([len(rPos), x+w+1]), y+1:np.min([len(rPos), y+2+w])][::-1])
quad_stats = []
for quad in [a11, a12, a21, a22]:
w_steps, diags = np.transpose(np.array([[j, np.diagonal(quad.toarray(), j).mean()] for j in np.arange(-w, w+1)]))
if np.var(diags) <= 0:
quad_stats.append(0)
else:
quad_stats.append(kendalltau(w_steps, diags).statistic)
tree_stats = (1 + np.array(quad_stats).reshape([2,2]))/2
return -np.linalg.det(tree_stats)
def detectButterfly(intensitySquare, symmetry=0):
partition_sum = intensitySquare.sum()
if partition_sum == 0:
normalized_square = [[0,0], [0,0]]
else:
normalized_square = intensitySquare / partition_sum
if symmetry == 0:
return [normalized_square[1,0], normalized_square[0,1]]
else:
return [normalized_square[0,0], normalized_square[1,1]]
def testButterflyStructure(mat, x, y, w, resamples=100):
submatrix = mat[np.max([0, x-w[0]]):np.min([len(rPos), x+w[0]+1]), np.max([0, y-w[1]]):np.min([len(rPos), y+w[1]+1])].toarray()
def formSubmatrices(matrix):
return matrix[:w[0], :w[1]], matrix[:w[0], w[1]+1:], matrix[w[0]+1:, :w[1]], matrix[w[0]+1:, w[1]+1:]
treeStat = np.mean([matrix.var() * matrix.size / (matrix.size - 1) for matrix in formSubmatrices(submatrix)])
permutedTreeStat = []
for _ in range(resamples):
n, m = submatrix.shape
permutedSubmatrix = submatrix[rng.permutation(n), :][:, rng.permutation(m)]
permutedTreeStat.append(np.mean([matrix.var() * matrix.size / (matrix.size - 1) for matrix in formSubmatrices(permutedSubmatrix)]))
return (permutedTreeStat > treeStat).mean()
def testButterflyCandidate(mat, point, ws, diagonalw=10, resamples=10, symmetry=0):
largeCandidate = pickLargeCandidate(mat, *point, ws)
chis = np.sort([xToChi(point[0]), xToChi(point[1])])
if symmetry == 0:
shift = np.array([1, 0])
intensity_indices = np.array([[0,0], [0,-1], [1,-1], [1,0], [-1,1], [-1,2], [-2,2], [-2,1]])
else:
shift = np.array([0, 0])
intensity_indices = np.array([[0,0], [0,-1], [-1,-1], [-1,0], [1,1], [1,2], [2,2], [2,1]])
candidate = findButterflySummary(hic, *largeCandidate, np.round(np.array(ws) / 2).astype(int), symmetry)
candidate = [candidate[0] + shift, candidate[1]]
structure = testButterflyStructure(mat, *candidate[0], ws, resamples)
treeStat = treeStatistic(mat, *candidate[0], diagonalw)
blockScan = scanBlock2(mat, *chis, [3,3])
blockScan = blockScan[blockScan > 10**(-10)]
intensity_ratio_a = np.mean([mat[*index] for index in (candidate[0] + intensity_indices)]) - np.mean(blockScan)
intensity_ratio_b = np.std(blockScan) * np.sqrt(len(blockScan) / (len(blockScan) - 1))
intensity_ratio = intensity_ratio_a / intensity_ratio_b
detected_butterfly = detectButterfly(computeIntensitySquare(mat, *candidate[0], ws), symmetry)
return [candidate[0][0], candidate[0][1], candidate[1], treeStat, structure, intensity_ratio, detected_butterfly[0], detected_butterfly[1]]
def calibrateButterflyTests(testMat, ratioThreshold=5, symmetry=0):
flatten_test_matrix = testMat[:,:,[2,3]].reshape(21*21,2)
flatten_test_matrix = flatten_test_matrix[(np.abs(flatten_test_matrix).mean(axis=1) > 0) & (flatten_test_matrix.mean(axis=1) != np.nan)]
thetas = np.transpose([flatten_test_matrix.mean(axis=0), flatten_test_matrix.std(axis=0) * np.sqrt(len(flatten_test_matrix) / (len(flatten_test_matrix)-1))])
calibrated_values = np.zeros((testMat.shape)[:2])
if symmetry == 0:
for j in range(len(testMat)):
for k in range(len(testMat)):
element = testMat[j, k]
if (np.array_equal(element, np.zeros(8))) or (element[5] < ratioThreshold):
calibrated_values[j,k] = 0
else:
a = 1-sp_stats.norm(loc=thetas[0,0], scale=thetas[0,1]).cdf(element[2])
b = 1-sp_stats.norm(loc=thetas[1,0], scale=thetas[1,1]).cdf(element[3])
c = element[4]
print(element)
calibrated_values[j,k] = np.min([a, b, c])
return calibrated_values
</code>
# Analysis
Each of the following sections performs parts of the analysis described in the main paper.
## Load Hi-C Matrix
Replace "hi-c_matrix" with filename of interest
<code>
row_indices, col_indices, hic_vals = np.transpose(np.loadtxt('hi-c_matrix'))
relevant_indices = (row_indices < 57509 - 1) & (col_indices < 57509 - 1)
hic = sp_sparse.coo_array((hic_vals[relevant_indices], (row_indices[relevant_indices].astype(int)-1, col_indices[relevant_indices].astype(int)-1)), (57509, 57509))
hic = sp_sparse.csr_array(hic)
del row_indices, col_indices, hic_vals, relevant_indices
</code>
## Copy Number & Mixture Proportion Inference
Estimation of effective copy number profile.
<code>
# Un-comment the respective lines, if the experimental protocol (Fix-C or Hi-C) of the contact map in question is known.
rowsums = covariateCorrection1(attachCovariates(hic.diagonal()))
# rowsums = dataCcorrector(hic.diagonal(), 'FixC')
# rowsums = dataCcorrector(hic.diagonal(), 'HiC')
</code>
<code>
pi1, f, ps = estimate_proportion_ploidy(rowsums, 5)
</code>
## Off-diagonal inference
Filter Hi-C matrix by criteria specified in covariate correction
<code>
filtered_hic = hic[rPos,:][:,rPos]
</code>
### Type (a) events
<code>
# Extract change point candidates
candidates = np.abs(np.diff(medianFilter(pi1, 50))).nonzero()[0]
candidates = candidates[candidates < corrchromlocations[-1]]
aCandidates = []
extended_candidates = np.concatenate([[0], candidates, [len(rPos)]])
for j in range(1, len(extended_candidates)-1):
if np.max([extended_candidates[j] - extended_candidates[j-1], extended_candidates[j+1] - extended_candidates[j]]) > 100:
aCandidates.append(extended_candidates[j])
del candidates, extended_candidates, j
</code>
<code>
# Generate row-wise null distributions for extracted change-point candidates
mat = filtered_hic + filtered_hic.transpose() - sp_sparse.diags([filtered_hic.diagonal()], [0])
ws = [300, 300]
testPoints = np.concatenate([aCandidates, acrox]).astype(int)
aRow = []
for testPoint in testPoints:
if testPoint < np.min(ws):
aRow.append(rowPattern(mat, testPoint, [testPoint, testPoint]))
else:
aRow.append(rowPattern(mat, testPoint, ws))
del mat, ws, testPoints, testPoint
</code>
<code>
# Calculate summary statistics for each pair of change-point candidates
mat = filtered_hic + filtered_hic.transpose() - sp_sparse.diags([filtered_hic.diagonal()], [0])
ws = [300, 300]
testPoints = np.concatenate([aCandidates, acrox]).astype(int)
aSummary = np.nan * np.ones([len(testPoints), len(testPoints), 3])
for k in range(len(testPoints)):
for j in range(len(testPoints)):
x = testPoints[k]
y = testPoints[j]
rowSamples = aRow[k]
columnSamples = aRow[j]
target = detectPattern(computeIntensitySquare(mat, x, y, ws))
if (x>=y) or (xToChi(x) >= xToChi(y)) or (np.max([len(testPoints) - k, len(testPoints) - j]) <= 3):
aSummary[k, j] = [0, 0, 0]
else:
aSummary[k, j] = [(rowSamples <= target).mean(), (columnSamples <= target).mean(), testTreeStructure(mat, x, y, ws, 100) ]
del mat, ws, testPoints, k ,j
</code>
<code>
# Aggregate summary statistics into well-calibrated p-values
testPoints = np.concatenate([aCandidates, acrox]).astype(int)
aP = np.nan * np.ones([len(testPoints), len(testPoints)])
for k in range(len(testPoints)):
for j in range(len(testPoints)):
if (k>=j) or (xToChi(testPoints[k]) >= xToChi(testPoints[j])) or (np.abs(testPoints[k] - testPoints[j])<2000):
aP[k,j] = 0
else:
aP[k,j] = np.min(aSummary[k,j])
del testPoints, k, j
</code>
### Type (b) events
<code>
# Extract candidate points
bCandidates = np.nan * np.ones([21, 19 + 2, 3])
for chi1 in range(1, 22):
for chi2 in range(1, 22):
if chi1 < chi2:
[(pt1, pt2), size] = extractButterflyCandidate(filtered_hic, chi1, chi2, [50,50], 1)[0]
bCandidates[chi1-1, chi2-1] = np.array([pt1, pt2, size])
del chi1, chi2, pt1, pt2, size
</code>
<code>
# Compute summary statistics
mat = filtered_hic
ws = [50, 50]
bP = np.nan * np.ones([2, 21, 21, 8])
for par in [0,1]:
for chi1 in range(1, 22):
for chi2 in range(1, 22):
if chi1 >= chi2:
bP[par, chi1-1, chi2-1] = np.zeros(8)
else:
bP[par, chi1-1, chi2-1] = testButterflyCandidate(mat, (corrchromlocations[[chi1-1, chi2-1]]+25+bCandidates[chi1-1, chi2-1, 1]).astype(int), ws, 10, 100, par)
del mat, ws, chi1, chi2, par
</code>
<code>
# Convert summaries into well-calibrated p-values
calibratedbP = np.array([calibrateButterflyTests(bP[0], 5, 0), calibrateButterflyTests(bP[1], 5, 1)])
</code>
<code>
# Report events corresponding to significant p-values
candidatesa = np.concatenate([aCandidates, acrox]).astype(int)
candidatesb = np.nan * np.ones([2, len(bP[0]), len(bP[0]), n, 5])
for par in [0,1]:
for j in range(len(bP[0])):
for k in range(len(bP[0])):
candidatesb[par, j, k] = np.array([bP[par, j, k, 0], bP[par, j, k, 1], xToChi(bP[par, j, k, 0]), xToChi(bP[par, j, k, 1]), calibratedbP[par, j, k]])
list = np.nan * np.ones([len(candidatesa), len(candidatesa), 5])
for j in range(len(candidatesa)):
for k in range(len(candidatesa)):
list[j,k] = np.array([candidatesa[j], candidatesa[k], xToChi(candidatesa[j]), xToChi(candidatesa[k]), aP[j,k]])
list = np.concatenate([np.concatenate(list), np.concatenate(np.concatenate(candidatesb[0])), np.concatenate(np.concatenate(candidatesb[1]))])
list = list[list[:,-1] > 0]
list = list[list[:,-1].argsort()][::-1]
threshold = findThreshold(list[:,-1], 2)
</code>
|
{
"filename": "HiDENSEC.ipynb",
"repository": "songlab-cal/HiDENSEC",
"query": "transformed_from_existing",
"size": 51034,
"sha": ""
}
|
# DESeq2_4.ipynb
Repository: LucaMenestrina/DEGA
# DESeq2 Use Case
## Load Libraries
<code>
library("DESeq2")
library("genefilter")
</code>
Set variables (data from the [Bottomly et al.](https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0017820) dataset)
<code>
GENE_COUNTS = "https://raw.githubusercontent.com/LucaMenestrina/DEGA/main/validation/bottomly_counts.csv" # "bottomly_counts.csv"
PHENO_DATA = "https://raw.githubusercontent.com/LucaMenestrina/DEGA/main/validation/bottomly_phenotypes.csv" # "bottomly_phenotypes.csv"
VAR_TO_TEST = "strain"
</code>
Load data
<code>
colData <- read.csv(PHENO_DATA, sep=",", row.names=1)
countData <- as.matrix(read.csv(GENE_COUNTS, row.names="X"))
# filter and sort countData columns on the basis of colData index
# (they have to be in the same order)
countData <- countData[, rownames(colData)]
</code>
Create DESeq2 object
<code>
dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = as.formula(paste("~", VAR_TO_TEST)))
</code>
Run the differential expression analysis
<code>
dds <- DESeq(dds)
res <- results(dds, alpha=0.05, lfcThreshold=0)
resS = lfcShrink(dds, alpha=0.05, lfcThreshold=0, coef=2, type="normal")
</code>
<code>
summary(resS, alpha=0.05)
</code>
<code>
# Save results
# write.csv(res, "DESeq2_bottomlyResults.csv")
# write.csv(resS, "DESeq2_bottomlyWithShrinkageResults.csv")
</code>
## References
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. _Genome Biology_, _15_(12), 550. [https://doi.org/10.1186/S13059-014-0550-8/FIGURES/9](https://doi.org/10.1186/S13059-014-0550-8/FIGURES/9)
Bottomly, D. et al. (2011). Evaluating Gene Expression in C57BL/6J and DBA/2J Mouse Striatum Using RNA-Seq and Microarrays. _PLOS ONE_, _6_(3), e17820. [https://doi.org/10.1371/JOURNAL.PONE.0017820](https://doi.org/10.1371/JOURNAL.PONE.0017820)
|
{
"filename": "DESeq2_4.ipynb",
"repository": "LucaMenestrina/DEGA",
"query": "transformed_from_existing",
"size": 14505,
"sha": ""
}
|
# Project_未命名.ipynb
Repository: Peevin/TNBC
<code>
import scanpy as sc
import pandas as pd
import numpy as np
</code>
<code>
sc.settings.set_figure_params(dpi=300, facecolor='white')
</code>
<code>
adata = sc.read_h5ad('/Users/liupeiwen/BC/21 CC Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer/data/cc.bc.sc.T.ann.h5ad')
</code>
<code>
adata.obs
</code>
<code>
np.unique(adata.obs['Sub_Cluster'])
</code>
<code>
adata.obs['label'] = np.where(adata.obs['Sub_Cluster']=='t_pDC-LILRA4','pDC',adata.obs['Global_Cluster'])
</code>
<code>
np.unique(adata.obs['label'])
</code>
<code>
sc.pp.normalize_total(adata,target_sum=1e6)
</code>
<code>
sc.pl.dotplot(adata,var_names=['TNFSF9','SIGLEC9'],groupby='label',save='.pdf')
</code>
|
{
"filename": "Project_未命名.ipynb",
"repository": "Peevin/TNBC",
"query": "transformed_from_existing",
"size": 255361,
"sha": ""
}
|
# taxonomy_explore_github_topics.ipynb
Repository: kuefmz/define
<code>
import pandas as pd
</code>
<code>
df = pd.read_csv('topics.csv')
</code>
<code>
df.head()
</code>
<code>
df.shape
</code>
<code>
print('Number of different topics on GitHub')
len(df['topic'].unique())
</code>
<code>
topic_counter = {}
for ind, row in df.iterrows():
if row['topic'] not in topic_counter.keys():
topic_counter[row['topic']] = row['num_pushers']
else:
topic_counter[row['topic']] += row['num_pushers']
</code>
<code>
print('Number of unique topics: ', len(topic_counter.keys()))
</code>
<code>
topic_counts = []
for k in topic_counter.keys():
#print(k, ' - ', topic_counter[k])
topic_counts.append((k, topic_counter[k]))
</code>
<code>
topic_counts.sort(key=lambda x: x[1], reverse=True)
</code>
<code>
topic_counts
</code>
|
{
"filename": "taxonomy_explore_github_topics.ipynb",
"repository": "kuefmz/define",
"query": "transformed_from_existing",
"size": 48165,
"sha": ""
}
|
# bioinformatics_bootcamp_2018_ATAC-seq-checkpoint_2.ipynb
Repository: ryanmarina/BMS
# BIOM 200 bioinformatics bootcamp - ATAC-seq analysis
* [(Pre-class) Introduction](#introduction)
* [(Pre-class) Installations](#installations)
* [(In-class) Data processing](#processing)
* [(In-class) Data analysis](#processing)
* [(In class) Genome browser](#genomebrowser)
* [(Optional) Single cell ATAC-seq](#scatac)
## <a name="introduction"></a>Introduction
ATAC-seq is an assay that captures accessible chromatin first described in [Buenrostro et al. 2013](https://doi.org/10.1038/nmeth.2688), and it stands for Assay for Transposase-Accessible Chromatin using sequencing. It has become a popular assay because of its advantages over previous assays (DNAseI-seq and FAIRE-seq), such as the relatively easy protocol and low cellular input.
In this tutorial, we will cover:
* Processing ATAC-seq data
* High level summary using gene ontology
* Finding enriched transcription factor motifs
* Visualizing results on the UCSC genome browser
___

___
Unlike something like ChIP-seq, ATAC-seq is typically not run with a control due to the limited information obtained. ATAC-seq is typically sequenced with paired-end sequencing for the following reasons:
* More sequence data leads to better alignment results. Many genomes contain numerous repetitive elements, and failing to align reads to certain genomic regions unambiguously renders those regions inaccessible to the assay.
* With ATAC-seq, we are interested in knowing the full span of the DNA fragments generated by the assay. A DNA fragment generated by the ATAC is typically longer than a sequence read, so a read will define only one end of the fragment. Therefore, with single-end sequencing, we would have to guess where the other end of the fragment is. Since paired-end sequencing generates reads from both ends, the full span of the DNA fragment is known precisely.
* PCR duplicates are identified more accurately. PCR duplicates are artifacts of the ATAC-seq procedure, and they should be removed as part of the analysis pipeline. However, computational programs that remove PCR duplicates (e.g. Picard's MarkDuplicates) typically identify duplicates based on comparing ends of aligned reads.
## <a name="installations"></a>Installations
### Command-line imports
TSCC has a few programs installed already, so all we have to do is load them. To check all available modules, you can type `module avail` on the command line. Add the following lines to your `.bashrc` file. These will automatically load the program [bwa](http://bio-bwa.sourceforge.net/bwa.shtml) and [bedtools](https://bedtools.readthedocs.io/en/latest/) each time you open a new terminal shell. We also need to tell bash to search in the following places for `trim_galore` and `homer`, which we will be using later.
```
PATH=$PATH:/oasis/tscc/scratch/biom200/bms_2018/programs
PATH=$PATH:/oasis/tscc/scratch/biom200/bms_2018/homer/bin
module load bwa
module load bedtools
```
Now open a new terminal window. Typing in `bwa` should bring up the user manual.
### Python installations
We will need to install a couple programs and modules.
```
conda install -c bioconda cutadapt macs2
```
### Index the reference genome for BWA
We will need an indexed reference genome for the BWA algorithm. Each aligner program has expects its own index, so we can't simply reuse the one we built for STAR in the RNA-seq alignment section. In the interest of time, I have already downloaded and prepared a reference genome for you. We will just need to link it over from the common directory.
```
ln -s /oasis/tscc/scratch/biom200/bms_2018/bwa_ref/ ~/scratch/bwa_ref
```
If you want to prepare the reference genome yourself, download the GRCm38 (mm10) genome sequence from gencode and use `bwa index` to prepare it.
```
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M18/GRCm38.p6.genome.fa.gz
gunzip GRCm38.p6.genome.fa.gz
mv GRCm38.p6.genome.fa mm10.fa
bwa index mm10.fa
```
## <a name="processing"></a>Data processing
### Obtaining the datasets
These ATAC-seq datasets are from the ENCODE project website and were generated from the same samples that were used for the RNA-seq portion of this bootcamp.
You can find them here:
https://www.encodeproject.org/experiments/ENCSR347RZI/
https://www.encodeproject.org/experiments/ENCSR984HFU/
To simplify things, we will start with processing the 4 hour timepoint.
Create a new directory for ATAC-seq and create links to the `.fastq` files that were pre-downloaded.
```
mkdir -p ~/raw_data/ATAC/ ~/projects/ATAC/
ln -s /oasis/tscc/scratch/biom200/bms_2018/atac_seq/raw_data/*.fastq.gz ~/raw_data/ATAC/
cd ~/projects/ATAC/
```
### Trimming adapter sequences from reads
Adapter trimming is necessary because of the way ATAC-seq works (tagmentation, where the DNA is simulaneously fragmented and sequencing adapters are added). Adapters in the sequencing data may prevent the reads from aligning to the reference genome and adversely affect the downstream analysis.
For ATAC-seq data, we trim adapter sequence using a program called [trim_galore](https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/Trim_Galore_User_Guide.md). The nice thing about this program is that it automatically detects which adapters are present in your reads (Nextera adapters for ATAC-seq).
We need to specify that our reads are paired-end (`--paired`). This could take a while, but it's not memory intensive. Submit this as a script (`qsub -V`).
```
#!/bin/bash
#PBS -q hotel
#PBS -N trim_adapters
#PBS -l nodes=1:ppn=4
#PBS -l walltime=1:00:00
#PBS -o trim_adapters.out
#PBS -e trim_adapters.err
trim_galore --paired --output_dir ~/projects/ATAC/ ~/raw_data/ATAC/dendritic.4_hour.R1.fastq.gz ~/raw_data/ATAC/dendritic.4_hour.R2.fastq.gz
```
The program will take a while to run, but in the end it should output two new files in the `~/projects/ATAC/` directory with trimmed reads:
`dendritic.4_hour.R1_val_1.fq.gz` and `dendritic.4_hour.R2_val_2.fq.gz`.
___
### Align reads to the reference genome
We can now align the reads to a reference genome (mm10) and look for peaks of transposition activity. We will use the [bwa mem](http://bio-bwa.sourceforge.net/bwa.shtml) aligner and pipe the output to [samtools](http://www.htslib.org/doc/samtools.html) for an initial filter on high quality mapped reads with `samtools view`. We will next use `samtools fixmate`, which is necessary for the duplicate removal step later. We will finally sort by coordinate with `samtools sort` and use `samtools markdup` to mark and remove duplicate sequences. In this long chain of commands, we use `-` to indicate we want to read the output from the previous command.
We will need a copy of the mm10 reference genome prepared earlier (see installations for instructions for preparing a different reference genome, e.g. hg19 for human data).
*WARNING*: This command may take a long time to run, so submit it as a script. Use `qsub -V` to ensure that your paths get copied over to the job.
```
#!/bin/bash
#PBS -q hotel
#PBS -N bwa_mapping
#PBS -l nodes=1:ppn=16
#PBS -l walltime=1:00:00
#PBS -o bwa_mapping.out
#PBS -e bwa_mapping.err
module load bwa
bwa mem -t 16 ~/scratch/bwa_ref/mm10.fa ~/raw_data/ATAC/dendritic.4_hour.R1_val_1.fq.gz ~/raw_data/ATAC/dendritic.4_hour.R2_val_2.fq.gz \
| samtools view -@ 8 -b -u -h -f 3 -F 256 -F 2048 -q 30 - \
| samtools fixmate -m - - \
| samtools sort -m 4G -@ 8 - \
| samtools markdup -r - ~/projects/ATAC/dendritic.4_hour.nodup.bam
```
We used a couple of argument flags for `samtools view`. Here are what some of them mean:
* `-b`: output BAM or binary format
* `-u`: output uncompressed which is useful for piping to other samtools commands
* `-h`: output header
* `-f 3`: only include alignments marked with the SAM flag `3`, which means "properly paired and mapped"
* `-F 256`: exclude alignments with flag `256`, which means that bwa mapped the read to multiple places in the reference genome, and this alignment is not the best
* `-F 2048`: exclude alignments marked with SAM flag `2048`, indicating chimeric alignments, where bwa decided that parts of the read mapped to different regions in the genome. These records are the individual aligned segments of the read. They usually indicate structural variation. We're not going to base peak calls on them.
* `-q 30`: exclude alignments with a mapping score < 30
The manual page for samtools has more information on flags and how to use them to filter your alignments.
It's hard to remember what the flags mean. Lucky for us, there's a web-based tool by the Broad Institute to [explain SAM flags](https://broadinstitute.github.io/picard/explain-flags.html).
___
### Call accessible chromatin peaks
Let's assume that we have aligned and processed both datasets (0 hour, 4 hour). We have the final `.bam` files from both of these datasets, so the next step is to call peaks on all of them.
We will next use [MACS2](https://github.com/taoliu/MACS) to call peaks (regions with high transposition activity) on the aligned reads. Use `qsub -V` to ensure that your paths get copied over to the job.
```
#!/bin/bash
#PBS -q hotel
#PBS -N peaks
#PBS -l nodes=1:ppn=1
#PBS -l walltime=1:00:00
#PBS -o peaks.out
#PBS -e peaks.err
macs2 callpeak -t ~/projects/ATAC/dendritic.4_hour.nodup.bam -n dendritic.4_hour --outdir ~/projects/ATAC/ -g mm --nomodel --shift -100 --extsize 200
```
MACS2 calls peaks using the default false discovery rate (FDR) of 0.05. If you don't know what an false discovery rate is, don't worry, you'll learn more about it in the statistics class in Winter/Spring quarter. Basically, instead of setting a hard p-value cutoff of 0.05 every time regardless of the p-value distribution, an FDR considers the shape of the p-value distribution and adjusts accordingly.
We used a couple of argument flags for `macs2 callpeak`. Here are some of them mean:
* `-t`: the "treatment" file -- the input, which is the BAM file from the previous step
* `-n`: the name of the experiment, which is used to name files
* `-g`: the genome's mappable size; 'mm' is an alias for the mouse genome's mappable size
* `--nomodel, --shift, and --extsize`: MACS2 was originally designed for ChIP-seq data, so we're telling it not to use its built-in model, but to extend and shift reads in a way appropriate for ATAC-seq.
When the program finishes running, there should be an output file called `dendritic.0_hour_peaks.narrowPeak`.
These are our final ATAC-seq peaks in [BED format](https://genome.ucsc.edu/FAQ/FAQformat.html), which is a file format that contains genomic coordinates.
Open the peaks file for viewing
```
less -S ~/projects/ATAC/dendritic.4_hour_peaks.narrowPeak
```
This should be a tab-separated BED file - each row in this file contains information about a single ATAC-seq peak. The first three columns will contain information about where the peak is located within the genome, specifically these are chromosome, start position, and end position.
>Here is an example of what you should see.

**Now run the same command on the other dataset.**
___
If you've reached this point, try processing the other dataset.
*HINT*: You can put everything together (trimming adapters, aligning, calling peaks) in a script and submit it as a job. Make sure you change the filenames!
## <a name="analysis"></a>Data analysis
### bedtools
[BEDtools](https://bedtools.readthedocs.io/en/latest/) (bedtools) is a command-line program that was designed around manipulating BED files. Try typing in `bedtools` on the command line and you will see that it has a wide range of functions.
We will use bedtools to see how many peak calls overlap and differ between our two timepoints.
Let's check the concordance between the peak calls.
We can first check how many lines are in each BED file with a simple `wc -l` (line count) command. **How many peaks were called for each timepoint?** Modify this command to count the lines for both BED files.
```
wc -l ~/projects/ATAC/dendritic.4_hour_peaks.narrowPeak
```
Next, we will use the `bedtools intersect` command to figure out how many peaks are common to both datasets.
You can find the documentation for it [here](https://bedtools.readthedocs.io/en/latest/content/tools/intersect.html).
**How would you use this command to find out the number of common peaks between the two timepoints?**
*HINT*: pipe your outputs to `wc -l`
Now that we know how many peaks are similar between the two timepoints, let's find out the number of peaks that differ between them.
**How would you use the bedtools intersect command to do this?**
*HINT*: you want the inverse of intersect
___
### GREAT (gene ontology for non-coding regions)
[GREAT](http://great.stanford.edu/public/html/) stands for Genomic Regions Enrichment of Annotations Tool, which is basically a fancy way to say gene ontology without using genes as input. Instead, the input is used-defined genomic regions, which could be from any range of genomic assays (e.g. ATAC-seq, ChIP-seq, WGBS). GREAT assigns genomic regions to genes using a combination of methods. Based on these gene assignments, it then runs standard gene ontology which can provide a high-level summary of the data.
Here, we will use GREAT to analyze the broad changes between the 4 hour and 0 hour timepoints. Specifically, we will see what gene ontology terms are enriched in the peaks specific to the 4 hour timepoint as compared to all 4 hour timepoint peaks.
First, we will want to format our `narrowPeak` files into test and background regions for input into GREAT.
GREAT expects a BED file as input with 4 columns: chromosome, start, end, and peak name.
Use the `bedtools intersect` command that we used earlier to find peaks specific to the 4 hour timepoint.
Our `narrowPeak` file contains some extraneous information that we don't need for GREAT, so we will use the `cut` utility to extract the columns we need (`-f 1-4` means columns 1-4).
```
bedtools intersect -a ~/projects/ATAC/dendritic.4_hour_peaks.narrowPeak \
-b ~/projects/ATAC/dendritic.0_hour_peaks.narrowPeak -v | cut -f 1-4 \
> ~/projects/ATAC/dendritic.4h_specific.bed
cut -f 1-4 ~/projects/ATAC/dendritic.4_hour_peaks.narrowPeak > ~/projects/ATAC/dendritic.4h_background.bed
```
Next, transfer the GREAT input files to a location on your local computer.
You can use `scp` (or drag + drop on the sidebar for MobaXterm users).
Navigate to the [GREAT web tool](http://great.stanford.edu/public/html/).
We will use these options:
**Species Assembly**: `Mouse: NCBI build 38 (UCSC mm10, Dec/2011)`
**Test regions**: `dendritic.4h_specific.bed`
**Background regions**: `dendritic.4h_background.bed`
It will take a few minutes, but the results should return in the same web browser.
Scroll down to MSigDB Pathway results. Do the results make sense?
> You can visualize the results as a bar graph. It should look something like this.

___
### Motif enrichment
One of the powerful uses of ATAC-seq is to find the transcriptional drivers that mediate changes in expression. Through a sequence motif enrichment analysis, we can use the DNA sequences underlying the accessible chromatin regions to discover which transcription factors (TFs) are likely driving these changes.
<img align='right' src='http://homer.ucsd.edu/homer/pic2.gif'>
We will use the program [HOMER](http://homer.ucsd.edu/homer/motif/), which was developed by [Chris Benner](https://profiles.ucsd.edu/christopher.benner) at UCSD. HOMER works by identifying sequences of various lengths that are enriched in the test set relative to the background. For the test set, we will use the 4h specific peaks, and we will use all 4h peaks as the background. HOMER can automatically create a GC-content matched background, but it's better to specify a background in this case because we want to know what TF motifs are enriched after treatment.
We will be using the `findMotifsGenome.pl` script from the HOMER program. You can find the documentation for this tool [here](http://homer.ucsd.edu/homer/ngs/peakMotifs.html).
```
#!/bin/bash
#PBS -q hotel
#PBS -N motifs
#PBS -l nodes=1:ppn=8
#PBS -l walltime=1:00:00
#PBS -o motifs.out
#PBS -e motifs.err
findMotifsGenome.pl ~/projects/ATAC/dendritic.4h_specific.bed mm10 homer_motifs -bg ~/projects/ATAC/dendritic.4h_background.bed -size 200 -nomotif -bits -mset vertebrates -p 8
```
The first three positional arguments to `findMotifsGenome.pl` are:
1. input BED file
2. HOMER reference genome
3. output directory
We used a couple of argument flags for `findMotifsGenome.pl`. Here are what some of them mean:
* `-bg`: specifies that we want to use a background regions file instead of having automatically match random regions
* `-size`: fragment size to use for motif finding
* `-nomotif`: skips the *de novo* motif scanning part of the HOMER routine (for speed)
* `-bits`: output the motif sequence logos scaled for information content
* `-mset`: use the vertebrates motif collection for known motif enrichment
This may take a while - HOMER will output a progress log.
When HOMER finishes running, there will be an output directory (`homer_motifs` which we specified earlier) containing all of the motif enrichment results. The file that contains the summary of the analysis is `knownResults.html`, which stores results for the top enriched motifs. Transfer this file to your local computer with `scp` (or drag + drop on the sidebar for MobaXterm users) and open it with your default web browser.
> You should see something that looks like this (this screenshot shows a truncated view of the top 5 results).

By far, the most enriched motif is NFKB, which makes sense given that LPS treatment should signal through Toll-like receptors and activate the NFKB pathway. However, among the top results we see examples of repeated known motif matches. This happens because the DNA binding motifs of TFs in the same family may be extremely similar, such as the OCT (POU) family (core sequence motif: ATGCAAAT).
## <a name="genomebrowser"></a>Genome browser
### Using pre-made genome browser tracks
I've compiled a UCSC genome browser session with some of the data we've been working with this bootcamp.
It often helps to visualize the data you're working with, instead of looking at numbers all the time.
https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=jchiou42&hgS_otherUserSessionName=BMS_bootcamp_2018
In this session, I have included the two replicates of RNA-seq data that Ryan used for differential expression, as well as the ATAC-seq data that I used.
You can use this session to visualize peaks in one dataset, but not the other.
For example, look up the region surrounding the Cd40 gene `chr2:165039617-165087673`, which has highly upregulated expression when dendritic cells are treated with LPS.

Although there are dramatic differences in expression, the chromatin profile only changes slightly (at the promoter and generally across the gene body).
___
### Making your own genome browser tracks (in the future...)
Usually ENCODE data will have `.bigWig` files that we can visualize in the UCSC genome browser. However in special cases where they don't have the tracks (such as the data that we've been using for this bootcamp), we will need to make the tracks ourselves.
First install [deeptools](https://deeptools.readthedocs.io/en/develop/), which is a program suite that has a lot of utilities for working with NGS data.
```conda install -c bioconda deeptools```
We will be using the `bamCoverage` script. This takes a `.bam` file as input and can output a `.bigWig` file, which is your genome browser signal track in binary format. Let's try this out on one of the RNA-seq bam files. We will need to first index the reads with `samtools`.
```
mkdir -p ~/projects/mouse_LPS/genome_browser
ln -s /oasis/tscc/scratch/biom200/bms_2018/rna_seq/analysis/star_alignment/bam_files/mouse_0hr_rep1_Aligned.sorted.bam ~/projects/mouse_LPS/genome_browser
samtools index ~/projects/mouse_LPS/genome_browser/mouse_0hr_rep1_Aligned.sorted.bam
bamCoverage --bam ~/projects/mouse_LPS/genome_browser/mouse_0hr_rep1_Aligned.sorted.bam --outFileName ~/projects/mouse_LPS/genome_browser/mouse_0hr_rep1.RNAseq.bw --binSize 50 --numberOfProcessors 1 --normalizeUsing RPKM --effectiveGenomeSize 1870000000 --skipNonCoveredRegions
```
Here the number 1870000000 refers to 1.87 gigabases, the approximate mappable genome size of the mouse genome mm10.
This should output the file `mouse_0hr_rep1.RNAseq.bw` in the `~/projects/mouse_LPS/genome_browser/` directory. We will need to upload this to an internet server with public access (here I'm using Amazon's web services (AWS) for this, TSCC should have an ftp directory that can face outwards).
However you choose to upload your file, you can now add it as a custom track in the UCSC Genome Browser using the bigDataUrl option.
```track type=bigWig name="Dend. 0hr rep1 LPS RNA" description="Dendritic 0hr rep1 LPS RNA signal" visibility=2 maxHeightPixels=64 db=mm10 color=128,128,128 bigDataUrl=https://s3-us-west-2.amazonaws.com/gaulton-lab-ucsc/public/BMS_bioinf_bootcamp/mouse_0hr_rep1.RNAseq.bw```
## <a name="scatac"></a>Single cell ATAC-seq
Single cell methods enable researchers to capture finer resolution than ever before, and allow pinpointing disease mechanisms and regulatory programs to specific cell types within a bulk tissue sample. The analysis of single cell ATAC-seq is many times more challenging than analyzing single cell RNA-seq, because accessible chromatin is essentially binary at the single-cell level.
___
For those interested in learning more about single cell ATAC-seq or those who have finished everything early, there is a well-documented tutorial for [analyzing a mouse atlas dataset](http://atlas.gs.washington.edu/mouse-atac/).
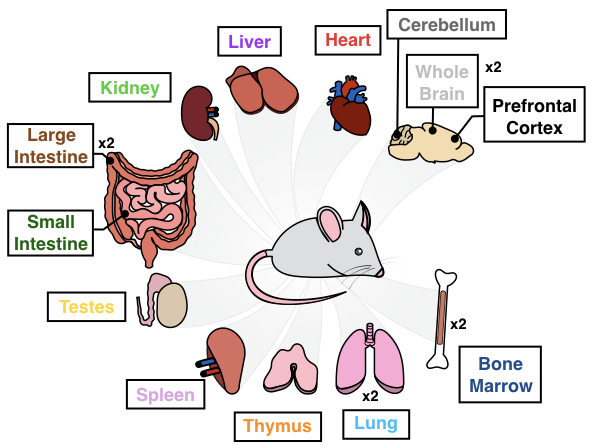
___
Papers that demonstrate use cases of single cell ATAC-seq include:
1. [Mouse Atlas](https://doi.org/10.1016/j.cell.2018.06.052)
2. [Fly Embryo](https://www.nature.com/articles/nature25981)
3. [Mouse Forebrain](https://www.nature.com/articles/s41593-018-0079-3)
4. [Hematopoietic Landscape](https://doi.org/10.1016/j.cell.2018.03.074)
|
{
"filename": "bioinformatics_bootcamp_2018_ATAC-seq-checkpoint_2.ipynb",
"repository": "ryanmarina/BMS",
"query": "transformed_from_existing",
"size": 28407,
"sha": ""
}
|
# thesis_homer_genome_annotation_1.ipynb
Repository: liouhy/2022-Charite-master
# HOMER - genome annotation
Here, we used HOMER to annotate genomic regions from scATAC-seq datasets. First, we created bed files of genomic regions.
<code>
import pandas as pd
import anndata as ad
</code>
<code>
# Granja et al.
ft = pd.read_csv('../raw/scATAC-Healthy-Hematopoiesis-191120-rows.csv')
ft_homer = pd.DataFrame()
ft_homer[['chr','start','end']] = ft.iloc[:,0].str.split('_',expand=True)
ft_homer['id'] = ft['name']
ft_homer['score'] = ft['score']
ft_homer['strand'] = '*'
ft_homer.to_csv('../raw/scATAC-Healthy-Hematopoiesis-191120-ft-homer.txt', sep='\t', header=None, index=None)
</code>
<code>
# Luecken et al.
r_adata = ad.read_h5ad("../processed/Luecken_multiome_BMMC-r_adata.h5ad")
index=r_adata.var.loc[r_adata.var['feature_types'] == 'ATAC',:].index
bed=index.str.split('-', expand=True).to_frame(index=False, name=['chr','start','end'])
bed.to_csv('../processed/Luecken_ATAC.bed', sep='\t', header=None, index=None)
</code>
<code>
# 10x
adata_atac = ad.read_h5ad('../processed/10X_multiome/pbmc_atac.h5ad')
bed = pd.DataFrame(adata_atac.var.index).iloc[:,0].str.split('-',expand=True)
bed.to_csv('../processed/10X_multiome/ATAC.bed', sep='\t', header=None, index=None)
</code>
Run the lines in homer_annotation.txt to perform annotation.
|
{
"filename": "thesis_homer_genome_annotation_1.ipynb",
"repository": "liouhy/2022-Charite-master",
"query": "transformed_from_existing",
"size": 3038,
"sha": ""
}
|
# table_model_1_1.ipynb
Repository: DongjoonLim/EvoLSTM
<code>
import numpy as np
from tqdm.notebook import tqdm
!nvidia-smi
</code>
<code>
k = 7
des = str(np.load('prepData/insert2Des__HPGPNRMPC_hg38_chr2.npy'))
anc = str(np.load('prepData/insert2Anc__HPGPNRMPC_hg38_chr2.npy'))
print(len(anc), len(des))
def buildTable(anc, des):
table = {}
freq = {}
for i in tqdm_notebook(range(len(anc)-k*2-1)):
try:
table[anc[i:i+k*2+1], des[i+k]] += 1
except KeyError:
table[anc[i:i+k*2+1], des[i+k]] = 1
try:
freq[anc[i:i+k*2+1]] += 1
except KeyError:
freq[anc[i:i+k*2+1]] = 1
for key in tqdm_notebook(table.keys()):
table[key] = table[key]/freq[key[0]]
return table, freq
table, freq = buildTable(anc,des)
print(table)
np.save('table_{}'.format(k*2+1), table)
</code>
<code>
ancName = '_HPGPNRMPC'
def load_seq(chromList):
inputAll = ''
predAll = ''
outputAll = ''
for chromosome in chromList:
try:
inputAll += str(np.load('prepData/insert2Anc_{}_hg38_chr{}.npy'.format(ancName,chromosome)))#[:10000000]
outputAll += str(np.load('prepData/insert2Des_{}_hg38_chr{}.npy'.format(ancName,chromosome)))#[:10000000]
predAll += str(np.load('prepData/simulated_{}_-1_chr{}.npy'.format(ancName, chromosome)))#[:10000000]
except FileNotFoundError:
print(chromosome)
continue
print(len(inputAll), len(outputAll), len(predAll))
print(inputAll[-10:], outputAll[-10:], predAll[-10:])
return [inputAll], [outputAll], [predAll]
inputAll, outputAll, predAll = load_seq([12,16,17,19,20,21,22])
</code>
<code>
np.set_printoptions(precision=15)
def model_simulate(alphabet, k, inputAll):
inputAll = inputAll[0]
table15 = np.load('table_15.npy',allow_pickle=True).item()
table11 = np.load('table_11.npy',allow_pickle=True).item()
table5 = np.load('table_5.npy',allow_pickle=True).item()
table1 = np.load('table_1.npy',allow_pickle=True).item()
pred_table = ''
for i in tqdm_notebook(range(len(inputAll))):
if i <k:
pred_table += inputAll[i]
elif k<=i<len(inputAll)-k:
mut_prob = []
for item in alphabet:
try:
mut_prob.append(table15[inputAll[i-7:i+7+1], item])
except KeyError:
try:
mut_prob.append(table11[inputAll[i-5:i+5+1], item])
except KeyError:
try:
mut_prob.append(table5[inputAll[i-2:i+2+1], item])
except KeyError:
try:
mut_prob.append(table1[inputAll[i], item])
except KeyError:
mut_prob.append(0)
# print(mut_prob)
# print(np.random.choice(len(mut_prob), 1, p=mut_prob))
# print(sum(mut_prob))
# mut_prob = [float(i)/sum(mut_prob) for i in mut_prob]
mut_prob = np.array(mut_prob)
mut_prob /= mut_prob.sum()
pred_table += alphabet[np.random.multinomial(1, mut_prob).argmax()]
# pred_table += alphabet[np.random.choice(len(mut_prob), 1, p=mut_prob)[0]]
else :
pred_table += inputAll[i]
return pred_table
</code>
<code>
# k = 7
mut_dict = np.load('mut_dict_insert2.npy',allow_pickle=True).item()
inv_dict = {v: k for k, v in mut_dict.items()}
print(list(inv_dict.keys()))
alphabet = list(inv_dict.keys())
# pred_table = model_simulate(alphabet, k, inputAll)
# np.save('predTable_{}'.format(k*2+1), pred_table)
# print(pred_table[:1000])
</code>
<code>
import math
def cross_entropy(alphabet, k, inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
table = np.load('table_{}.npy'.format(k*2+1),allow_pickle=True).item()
result = 0
count = 0
for i in tqdm_notebook(range(len(inputAll))):
if k<=i<len(inputAll)-k:
try:
result += -math.log(table[inputAll[i-k:i+k+1], outputAll[i]])
count +=1
except KeyError:
continue
# print('keyError')
return result/count
def cross_entropy39(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 19<=i<len(inputAll)-19:
try:
result += -math.log(table39[inputAll[i-19:i+19+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table21[inputAll[i-10:i+10+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table15[inputAll[i-7:i+7+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table11[inputAll[i-5:i+5+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table5[inputAll[i-2:i+2+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
def cross_entropy21(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 15<=i<len(inputAll)-10:
try:
result += -math.log(table21[inputAll[i-10:i+10+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table15[inputAll[i-7:i+7+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table11[inputAll[i-5:i+5+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table5[inputAll[i-2:i+2+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
def cross_entropy15(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 15<=i<len(inputAll)-7:
try:
result += -math.log(table15[inputAll[i-7:i+7+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table11[inputAll[i-5:i+5+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table5[inputAll[i-2:i+2+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
def cross_entropy11(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 15<=i<len(inputAll)-5:
try:
result += -math.log(table11[inputAll[i-5:i+5+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table5[inputAll[i-2:i+2+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
def cross_entropy5(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 15<=i<len(inputAll)-2:
try:
result += -math.log(table11[inputAll[i-5:i+5+1], outputAll[i]])
count +=1
except KeyError:
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
def cross_entropy1(alphabet,inputAll, outputAll):
inputAll = inputAll[0]
outputAll = outputAll[0]
result = 0
count = 0
for i in tqdm(range(len(inputAll))):
if 15<=i<len(inputAll):
try:
result += -math.log(table1[inputAll[i], outputAll[i]])
count +=1
except KeyError:
result += -math.log(0.01020408163265306)
count +1
print(result/count)
# print('keyError')
return result/count
</code>
<code>
table39 = np.load('table_39.npy',allow_pickle=True).item()
table21 = np.load('table_21.npy',allow_pickle=True).item()
table15 = np.load('table_15.npy',allow_pickle=True).item()
table11 = np.load('table_11.npy',allow_pickle=True).item()
table5 = np.load('table_5.npy',allow_pickle=True).item()
table1 = np.load('table_1.npy',allow_pickle=True).item()
print(len(table1.keys()))
print(1/98)
</code>
<code>
cross_entropy39(alphabet,inputAll, outputAll)
</code>
<code>
cross_entropy21(alphabet,inputAll, outputAll)
</code>
<code>
cross_entropy15(alphabet, inputAll, outputAll)
</code>
<code>
cross_entropy11(alphabet,inputAll, outputAll)
</code>
<code>
cross_entropy5(alphabet, inputAll, outputAll)
</code>
<code>
cross_entropy1(alphabet, inputAll, outputAll)
</code>
<code>
0.22000798713506123 0.20882172435559532 0.07722072727808955 0.098972098412289 0.11081867999770367 0.06958409766111845
</code>
<code>
cross_entropy2(alphabet, 0, inputAll, outputAll)
cross_entropy2(alphabet, 2, inputAll, outputAll)
cross_entropy2(alphabet, 5, inputAll, outputAll)
cross_entropy2(alphabet, 7, inputAll, outputAll)
cross_entropy2(alphabet, 10, inputAll, outputAll)
cross_entropy2(alphabet, 19, inputAll, outputAll)
</code>
|
{
"filename": "table_model_1_1.ipynb",
"repository": "DongjoonLim/EvoLSTM",
"query": "transformed_from_existing",
"size": 32967,
"sha": ""
}
|
# Evaluate_Integration_LISI.ipynb
Repository: pughlab/cancer-scrna-integration
---
# Evaluate data integration using LISI
*L.Richards*
*2021-06-14*
*/cluster/projects/pughlab/projects/cancer_scrna_integration/evalutation/lisi*
---
https://github.com/immunogenomics/LISI
<code>
# install.packages("devtools")
# devtools::install_github("immunogenomics/lisi")
library(lisi) # v1.0
library(Seurat)
library(rlist)
</code>
<code>
# list metadata files with embeddings (used for Fig 1 plotting)
embeddings.path <- "/cluster/projects/pughlab/projects/cancer_scrna_integration/figures"
embeddings <- list.files(embeddings.path, pattern = ".csv")
results <- list()
for (i in 1:length(embeddings)){
# load data
print(embeddings[i])
dat <- read.csv(paste0(embeddings.path, "/", embeddings[i]))
rownames(dat) <- dat$X
#####################
# calculate lisi for each method
methods <- unique(dat$Method)
for (j in 1:length(methods)){
print(methods[j])
# set up lisi input files
sub <- dat[dat$Method == methods[j], ] # subset methods
X <- sub[ ,c("Coords_1", "Coords_2")] # subset out embeddings
# calcualte and normalize lisi for samples and patients
lisi <- compute_lisi(X, sub, c("SampleID", "PatientID"))
lisi$SampleID_Norm <- lisi$SampleID / length(unique(sub$SampleID))
lisi$PatientID_Norm <- lisi$SampleID / length(unique(sub$PatientID))
#####################
# calcualte lisi for each cell type
# have to subset the dataframe by each cell type and calc
celltypes <- unique(sub$CellType)
lisi.celltypes <- list()
for (k in 1:length(celltypes)){
print(celltypes[k])
# subset embeddings by cell type
sub.cell <- sub[sub$CellType == celltypes[k], ]
X.cell <- X[rownames(sub.cell), ]
# calculate and normalize lisi within cell type
if(nrow(X.cell) < 40){
per <- 10
} else { per <- 30 }
print(per)
cell.lisi <- compute_lisi(X.cell, sub.cell, perplexity = per, c("SampleID", "PatientID"))
cell.lisi$SampleID_Norm <- cell.lisi$SampleID / length(unique(sub.cell$SampleID))
cell.lisi$PatientID_Norm <- cell.lisi$SampleID / length(unique(sub.cell$PatientID))
colnames(cell.lisi) <- paste0("CellType_", colnames(cell.lisi))
cell.lisi$CellType <- celltypes[k]
lisi.celltypes[[k]] <- cell.lisi
}
lisi.celltypes <- do.call(rbind, lisi.celltypes) # combine
lisi.celltypes <- lisi.celltypes[rownames(lisi), ] # reorder
identical(rownames(lisi.celltypes), rownames(lisi)) # sanity check
lisi <- cbind(lisi, lisi.celltypes) # combine cell and batch lisi
lisi$Method <- methods[j]
lisi$Study <- gsub("_MergedMeta.csv", "", embeddings[i])
colnames(lisi)[grep("_", colnames(lisi))] <- paste0("LISI_", colnames(lisi)[grep("_", colnames(lisi))])
results <- list.append(results, lisi)
}
}
results <- do.call(rbind, results) # combine across studies
dim(results) # 974206 rows
colnames(results)[1:2] <- paste0("LISI_", colnames(results)[1:2])
results$CellBarcode <- rownames(results)
</code>
<code>
# save results
write.csv(results, file = "LISI_calculations.csv")
</code>
|
{
"filename": "Evaluate_Integration_LISI.ipynb",
"repository": "pughlab/cancer-scrna-integration",
"query": "transformed_from_existing",
"size": 5486,
"sha": ""
}
|
# MCAsubset-checkpoint.ipynb
Repository: CSUBioGroup/scNCL-release
<code>
%load_ext autoreload
%autoreload 2
import os
import h5py
import seaborn as sns
import numpy as np
import pandas as pd
import scanpy as sc
import anndata
import csv
import gzip
import scipy.io
import scipy.sparse as sps
import matplotlib.pyplot as plt
from os.path import join
from sklearn.decomposition import PCA, IncrementalPCA
from sklearn.preprocessing import normalize
os.environ["CUDA_VISIBLE_DEVICES"] = "0"
np.random.seed(1234)
sc.settings.verbosity = 3
sc.logging.print_header()
</code>
## experiment id
<code>
exp_id = 'MCA-subset' + '-1301' # dataset_name + timestamp
</code>
## loading data
<code>
data_root = '/home/yxh/data/MCA/scjoint/atlas_subset'
adata_atac = sc.read_h5ad(join(data_root, 'adata_atac_cache.h5ad'))
adata_rna = sc.read_h5ad('/home/yxh/gitrepo/multi-omics/Portal-main/cache/adata_rna_facs.h5ad')
adata_rna.obs['domain'] = 'rna'
adata_atac.obs['domain'] = 'atac'
meta_rna = adata_rna.obs.copy()
meta_atac = adata_atac.obs.copy()
meta = pd.concat([meta_rna, meta_atac], axis=0)
</code>
<code>
adata_rna, adata_atac
</code>
<code>
# low-dimension representations of raw scATAC-seq data
atac_raw_emb = np.load('../cache/MCAsubset_atac_tsne.npy')
atac_raw_emb.shape
</code>
<code>
# params dict of preprocessing
ppd = {'binz': True,
'hvg_num':adata_atac.shape[1],
'lognorm':False,
'scale_per_batch':False,
'batch_label': 'domain',
'type_label': 'cell_type',
'knn': 10,
'knn_by_tissue':False
} # default settings
# outputs folder
output_dir = join(f'../outputs/{exp_id}')
os.makedirs(output_dir, exist_ok=True)
</code>
## model
<code>
import sys
sys.path = ["../"] + sys.path
import src.scNCL as scNCL
import src.utils as utls
</code>
<code>
model = scNCL.scNCL(
'non_linear', n_latent=64, bn=False, dr=0.2,
cont_w=0.05, cont_tau=0.4,
)
model.preprocess(
[adata_rna, adata_atac], # list of 'anndata' object
atac_raw_emb,
adata_adt_inputs=None,
pp_dict = ppd
)
if 1:
model.train(
opt='adam',
batch_size=500, training_steps=1000,
lr=0.001, weight_decay=5e-4,
log_step=50, eval_atac=False, #eval_top_k=1, eval_open=True,
)
else:
# loading checkpoints
ckpt_path = join(output_dir, 'ckpt_1000.pth')
model.load_ckpt(ckpt_path)
</code>
<code>
model.eval(inplace=True)
atac_pred_type = model.annotate()
# saving model
scNCL.save_ckpts(output_dir, model, step=1000)
</code>
<code>
ad_atac = sc.AnnData(model.feat_B)
ad_atac.obs = meta_atac.copy()
ad_atac.obs['pred_type'] = atac_pred_type
ad_atac.obs['pred_conf'] = np.max(model.head_B, axis=1)
</code>
<code>
ad_atac = utls.umap_for_adata(ad_atac)
</code>
<code>
sc.pl.umap(ad_atac, color=['cell_type', 'pred_type', 'pred_conf'])
</code>
# Evaluation
<code>
from src.metrics import osr_evaluator
share_mask = meta_atac.cell_type.isin(meta_rna.cell_type.unique()).to_numpy()
open_score = 1 - np.max(model.head_B, axis=1) # pb_max, logit_max_B
kn_data_pr = atac_pred_type[share_mask]
kn_data_gt = meta_atac.cell_type[share_mask].to_numpy()
kn_data_open_score = open_score[share_mask]
unk_data_open_score = open_score[np.logical_not(share_mask)]
closed_acc, os_auroc, os_aupr, oscr = osr_evaluator(kn_data_pr, kn_data_gt, kn_data_open_score, unk_data_open_score)
</code>
<code>
from sklearn.metrics import confusion_matrix
import matplotlib.pyplot as plt
import seaborn as sns
cm = confusion_matrix(meta_atac.cell_type.to_numpy(), atac_pred_type)
cm = cm/cm.sum(axis=1, keepdims=True)
df_cm = pd.DataFrame(cm, index = meta_atac.cell_type.unique(),
columns = meta_atac.cell_type.unique())
plt.figure(figsize = (10,7))
sns.heatmap(df_cm, )
</code>
|
{
"filename": "MCAsubset-checkpoint.ipynb",
"repository": "CSUBioGroup/scNCL-release",
"query": "transformed_from_existing",
"size": 199827,
"sha": ""
}
|
# GPT_1.ipynb
Repository: ZubairQazi/NDE-GPT
# GPT for Topic Categorization
<code>
import json
import pandas as pd
import numpy as np
import ast
import os
import re
from bs4 import BeautifulSoup
import csv
from tqdm.notebook import tqdm
import openai
from langchain.llms import OpenAI
from langchain.chat_models import ChatOpenAI
from langchain.callbacks import get_openai_callback
from langchain.schema.messages import HumanMessage, SystemMessage
</code>
## Load Data
EDAM topics, prompt, etc
<code>
with open("config.json", "r") as config_file:
config = json.load(config_file)
</code>
<code>
openai_api_key = config["api_keys"]["openai"]
</code>
<code>
dataset = pd.read_csv(input("Enter testing dataset path: "))
</code>
<code>
dataset['MeSH Terms'] = dataset['MeSH Terms'].apply(lambda mesh_list: np.unique(ast.literal_eval((mesh_list))))
dataset['EDAM Topics'] = dataset['EDAM Topics'].apply(lambda edam_list: np.unique(ast.literal_eval((edam_list))))
</code>
<code>
with open('templates/prompt_template.txt', 'r') as template_file:
template = template_file.read()
</code>
<code>
with open(input("Enter EDAM topics file:"), 'r') as edam_file:
full_edam_topics = edam_file.readlines()
full_edam_topics = [topic.strip() for topic in full_edam_topics]
</code>
<code>
# Add EDAM topics to prompt template
formatted_topics = "\n".join(full_edam_topics)
template = template.replace("<topics>", formatted_topics)
</code>
### Plots
Remove non-unique terms from each row's EDAM topic list
<code>
import matplotlib.pyplot as plt
# Create a histogram
plt.hist(dataset['EDAM Topics'].apply(len), bins='auto', edgecolor='black', alpha=0.7)
plt.hist(dataset['MeSH Terms'].apply(len), bins='auto', edgecolor='black', alpha=0.7)
plt.xlabel('Number of Topics / Terms')
plt.ylabel('Frequency')
plt.legend(['EDAM', 'MeSH'])
</code>
<code>
print("Total MeSH Terms:", len(list(dataset.iloc[0]['MeSH Terms'])))
print("Unique MeSH Terms:", len(np.unique(list(dataset.iloc[0]['MeSH Terms']))))
print()
print("Total EDAM Topics:", len(list(dataset.iloc[0]['EDAM Topics'])))
print("Unique EDAM Topics:", len(np.unique(list(dataset.iloc[0]['EDAM Topics']))))
</code>
### Remove any topics not in the EDAM Topics list
<code>
dataset['Filtered EDAM'] = dataset['EDAM Topics'].apply(lambda x: [item for item in x if item in full_edam_topics])
</code>
<code>
import matplotlib.pyplot as plt
# Create a histogram
plt.hist(dataset['EDAM Topics'].apply(len), bins='auto', edgecolor='black', alpha=0.7)
plt.hist(dataset['Filtered EDAM'].apply(len), bins='auto', edgecolor='black', alpha=0.7)
plt.xlabel('Number of Topics / Terms')
plt.ylabel('Frequency')
plt.legend(['EDAM', 'Filtered EDAM'])
</code>
<code>
# Check for any issues during filtering (missed topics, etc)
indices_true = dataset.loc[dataset['Filtered EDAM'].apply(lambda edam_list: not all(term in full_edam_topics for term in edam_list))].index
for index in indices_true:
edam_list = dataset.loc[index, 'Filtered EDAM']
terms_not_in_edam_topics = [term for term in edam_list if term not in full_edam_topics]
print(f"Index {index}: Terms not in edam_topics: {terms_not_in_edam_topics}")
</code>
## OpenAI API
Let's start with a proof of concept:
<code>
random_sample = dataset.sample(n=1)
index = random_sample.index[0]
description, abstract, paper_edam_topics = random_sample[['Description', 'Abstract', 'Filtered EDAM']].values[0]
</code>
<code>
prompt = template.replace('<abstract>', abstract).replace('<num_terms>', str(len(paper_edam_topics)))
# prompt = template.replace('<description>', description).replace('<num_terms>', str(len(paper_edam_topics)))
</code>
<code>
print(prompt)
</code>
<code>
messages = [
SystemMessage(content=f"You're a helpful assistant."),
HumanMessage(content=prompt)
]
</code>
<code>
chat = ChatOpenAI(
model_name='gpt-3.5-turbo',
openai_api_key = openai_api_key
)
gpt_output = ''
with get_openai_callback() as cb:
chat.invoke(messages)
for chunk in chat.stream(messages):
print(chunk.content, end="", flush=True)
gpt_output += chunk.content
print("\n===========CALLBACK: ==========\n")
print(cb)
print("\n=============\n")
# chat = OpenAI(
# model_name='text-davinci-003',
# openai_api_key = openai_api_key,
# temperature=0.75
# )
# gpt_output = ''
# with get_openai_callback() as cb:
# chat.invoke(messages)
# for chunk in chat.stream(messages):
# print(chunk, end="", flush=True)
# gpt_output += chunk
# print("\n===========CALLBACK: ==========\n")
# print(cb)
# print("\n=============\n")
</code>
<code>
gpt_output
</code>
<code>
not_in_edam = 0
true_topics = dataset.iloc[index]['Filtered EDAM']
num_correct = 0
for topic in gpt_output.strip().split(', '):
if topic not in full_edam_topics:
print(topic)
not_in_edam += 1
continue
if topic in true_topics:
num_correct += 1
</code>
<code>
print("GPT-outputted topics not in EDAM:", not_in_edam)
print("# Correct topics from GPT:", num_correct)
print("# Incorrect topics from GPT:", len(true_topics) - num_correct)
</code>
<code>
print(gpt_output, '\n')
print(', '.join(true_topics))
</code>
### Note: It seems as though GPT is not able to capture relevant topics given the entire list.
## Use GPT to capture major subtopics (Biology, Medicine, etc.)
We will see if GPT can capture the general topics of each data entry. Then we can pass the relevant subtopics.
https://bioportal.bioontology.org/ontologies/EDAM/?p=classes&conceptid=http%3A%2F%2Fedamontology.org%2Ftopic_0003
Biosciences - 4019
Chemistry - 3314
Computer science - 3316
Data management - 3071
Environmental Sciences - 3855
Informatics - 0605
Open science - 4010
Physics - 3318
<code>
import pandas as pd
edam_data = pd.read_csv("EDAM/EDAM.csv")
edam_data = edam_data[edam_data['Parents'].str.contains("http://edamontology.org/topic_")]
edam_data['Parents #'] = edam_data['Parents'].str.extractall(r'topic_(\d+)').groupby(level=0).agg(lambda parents: parents.tolist())
edam_data['Topic #'] = edam_data['Class ID'].apply(lambda url: url.split('topic_')[1])
</code>
<code>
from collections import defaultdict
topics = ['4019', '3314', '3316', '3071', '3855', '0605', '4010', '3318', '3361', '3068', '3678', '3315']
subtopics = defaultdict(lambda: [])
def get_children_topics(parent_id):
# children_ids = edam_data[edam_data['Parents'].str.contains(parent_id)]['Class ID'].apply(lambda url: url.split('topic_')[1]).to_list()
children_ids = edam_data[edam_data['Parents #'].apply(\
lambda parent_ids: parent_id in parent_ids)]['Topic #'].to_list()
if not len(children_ids):
return []
# print(parent_id, children_ids)
grandchildren = []
for child_id in children_ids:
grandchildren.append(get_children_topics(child_id))
children_ids.append(grandchildren)
return children_ids
for parent_topic in topics:
subtopics[parent_topic] = get_children_topics(parent_topic)
</code>
<code>
def flatten_list(nested_list):
flattened = []
for item in nested_list:
if isinstance(item, list):
flattened.extend(flatten_list(item))
else:
flattened.append(item)
return flattened
# Flatten each value in the dictionary
subtopics = {key: flatten_list(value) for key, value in subtopics.items()}
print()
for key, value in subtopics.items():
print(f"{key}: {value}")
</code>
<code>
topics_to_remove = ['3361', '3068', '3678', '3315']
topics_to_keep = ['4019', '3314', '3316', '3071', '3855', '0605', '4010', '3318']
for key in ['3361', '3068', '3678', '3315']:
topics_to_remove.extend(subtopics[key])
for key in ['4019', '3314', '3316', '3071', '3855', '0605', '4010', '3318']:
topics_to_keep.extend(subtopics[key])
topics_to_remove, topics_to_keep = set(topics_to_remove), set(topics_to_keep)
print('Number of Topics to remove: ', len([item for item in topics_to_remove if item not in topics_to_keep]))
edam_data = edam_data[~edam_data['Topic #'].apply(lambda topic: topic in topics_to_remove and topic not in topics_to_keep)]
# Remove the unnecessary topics =
for topic in ['3361', '3068', '3678', '3315']:
del subtopics[topic]
print()
for key, value in subtopics.items():
print(f"{key}: {value}")
</code>
<code>
parent_topics = defaultdict(lambda: set())
for key, values in subtopics.items():
for value in values:
parent_topics[value].add(key)
for key, value in parent_topics.items():
print(f"{key}: {value}")
</code>
<code>
main_topics = {}
for topic in ['4019', '3314', '3316', '3071', '3855', '0605', '4010', '3318']:
main_topics[topic] = edam_data[edam_data['Topic #'] == topic]['Preferred Label'].values[0]
main_topics
</code>
<code>
edam_data['Parent Topics'] = edam_data['Topic #'].apply(lambda topic:[main_topics[parent_topic] for parent_topic in parent_topics[topic]])
edam_data['Parent Topics']
</code>
<code>
dataset['Topic Category'] = dataset['Filtered EDAM'].apply(lambda edam_list: [edam_data[edam_data['Preferred Label'] == topic]['Parent Topics'].values[0] for topic in edam_list])\
.apply(lambda parent_list: set([item for sublist in parent_list for item in sublist]))
dataset['Topic Category']
</code>
## GPT for topic categories
<code>
with open('templates/prompt_template.txt', 'r') as template_file:
template = template_file.read()
formatted_topics = "\n".join(main_topics.values())
template = template.replace("<topics>", formatted_topics)
</code>
<code>
def get_accuracy(generated_topics, true_topics):
num_correct = 0
for topic in generated_topics.strip().split(', '):
if topic in true_topics:
num_correct += 1
return num_correct / len(true_topics)
</code>
<code>
def test(chat, dataset, truth_column='Topic Category', iterations=10, seed=54):
cost, accuracies = 0, []
io_pairs = []
random_samples = dataset.sample(n=iterations, random_state=seed)
for idx, random_sample in tqdm(random_samples.iterrows()):
index = random_sample.index[0]
_, abstract, paper_edam_topics = random_sample.loc[['Description', 'Abstract', truth_column]]
prompt = template.replace('<abstract>', abstract).replace('<num_terms>', str(len(paper_edam_topics)))
messages = [
SystemMessage(content=f"Generate a comma-separated list of relevant EDAM topics based on the provided abstract and topic categories."),
HumanMessage(content=prompt)
]
gpt_output = ''
with get_openai_callback() as cb:
chat.invoke(messages)
for chunk in chat.stream(messages):
if type(chat) == ChatOpenAI:
gpt_output += chunk.content
elif type(chat) == OpenAI:
gpt_output += chunk
cost += float(str(cb).split('$')[1])
try:
true_topics = dataset.iloc[index][truth_column]
accuracies.append(get_accuracy(gpt_output, true_topics))
except:
print('Error encountered at index', index)
io_pairs.append([abstract, ', '.join(true_topics), gpt_output])
print('Average Accuracy:', np.mean(accuracies))
print('Total Cost ($):', cost)
return accuracies, cost, io_pairs
</code>
<code>
chat = ChatOpenAI(
model_name='gpt-3.5-turbo',
openai_api_key = openai_api_key
)
accuracies, cost, _ = test(chat, dataset, iterations=50)
</code>
<code>
chat = OpenAI(
model_name='text-davinci-003',
openai_api_key = openai_api_key
)
accuracies, cost, _ = test(chat, dataset, iterations=50)
</code>
<code>
chat = ChatOpenAI(
model_name='gpt-4',
openai_api_key = openai_api_key
)
accuracies, cost, _ = test(chat, dataset, iterations=50)
</code>
## Double num_topics
Attempting to see if GPT will eventually get the correct topics
<code>
random_sample = dataset.sample(n=1)
index = random_sample.index[0]
description, abstract, paper_edam_topics = random_sample[['Description', 'Abstract', 'Filtered EDAM']].values[0]
</code>
<code>
prompt = template.replace('<abstract>', abstract).replace('<num_terms>', str(len(paper_edam_topics) * 2))
# prompt = template.replace('<description>', description).replace('<num_terms>', str(len(paper_edam_topics)))
</code>
<code>
print(prompt)
</code>
<code>
messages = [
SystemMessage(content=f"You're a helpful assistant."),
HumanMessage(content=prompt)
]
</code>
<code>
chat = ChatOpenAI(
model_name='gpt-3.5-turbo',
openai_api_key = openai_api_key
)
gpt_output = ''
with get_openai_callback() as cb:
chat.invoke(messages)
for chunk in chat.stream(messages):
print(chunk.content, end="", flush=True)
gpt_output += chunk.content
print("\n===========CALLBACK: ==========\n")
print(cb)
print("\n=============\n")
</code>
<code>
get_accuracy(gpt_output, paper_edam_topics)
</code>
<code>
print(abstract, '\n')
print(gpt_output)
print(', '.join(random_sample['Filtered EDAM'].values[0]))
</code>
There seems to be some potential, but requires further exploration. The main question to focus on is the definition of success, and how to measure whether the predicted topics are valid.
## Fine-Tuning GPT
We will start with 50 training samples for fine-tuning. (https://platform.openai.com/docs/guides/fine-tuning/preparing-your-dataset)
<code>
n = 100
training_data = dataset.sample(n=n, replace=False, random_state=50)
training_data.shape
</code>
<code>
with open(f"datasets/finetune-data-{n}.jsonl", 'w') as file:
for idx, row in training_data.iterrows():
description, abstract, paper_edam_topics = row[['Description', 'Abstract', 'Filtered EDAM']]
prompt = template.replace('<abstract>', abstract).replace('<num_terms>', str(len(paper_edam_topics)))
json_data = {
"messages": [
{"role": "system", "content": "Generate a comma-separated list of relevant EDAM topics based on the provided abstract and topic categories."},
{"role": "user", "content": prompt},
{"role": "assistant", "content": ', '.join(paper_edam_topics)}
]
}
file.write(json.dumps(json_data))
file.write('\n')
</code>
Use ```Chat_finetuning_data_prep.ipynb``` to check for any errors in the data and to get the cost estimate.
<code>
client = openai.OpenAI(api_key=openai_api_key)
fileobj = client.files.create(
file=open(f"datasets/finetune-data-{n}.jsonl", "rb"),
purpose="fine-tune"
)
fileobj
</code>
<code>
ftjob = client.fine_tuning.jobs.create(
training_file=fileobj.id,
model="gpt-3.5-turbo-1106"
)
ftjob
</code>
<code>
# Finished once fine_tuned_model is not None
client.fine_tuning.jobs.list(limit=10).data
</code>
<code>
# Most recent job
ftjob = client.fine_tuning.jobs.list(limit=10).data[0]
</code>
<code>
from io import BytesIO
training_results = pd.read_csv(BytesIO(client.files.content(ftjob.result_files[0]).content))[['step', 'train_loss', 'train_accuracy']]
training_results
</code>
<code>
training_results['train_loss'].plot()
plt.title('Loss Plot')
plt.xlabel('Step')
plt.ylabel('Loss')
</code>
<code>
training_results['train_accuracy'].plot()
plt.title('Accuracy Plot')
plt.xlabel('Step')
plt.ylabel('Accuracy')
</code>
## Testing All Models
Gather data for all the models
<code>
def test(chat, dataset, truth_column='Filtered EDAM', iterations=10, seed=54):
cost, accuracies = 0, []
io_pairs = []
random_samples = dataset.sample(n=iterations, random_state=seed)
for idx, random_sample in tqdm(random_samples.iterrows(), total=random_samples.shape[0]):
abstract, paper_edam_topics = random_sample.loc[['Abstract', truth_column]]
prompt = template.replace('<abstract>', abstract).replace('<num_terms>', str(len(paper_edam_topics)))
messages = [
SystemMessage(content=f"Generate a comma-separated list of relevant EDAM topics based on the provided abstract and topic categories."),
HumanMessage(content=prompt)
]
gpt_output = ''
try:
with get_openai_callback() as cb:
chat.invoke(messages)
for chunk in chat.stream(messages):
if type(chat) == ChatOpenAI:
gpt_output += chunk.content
elif type(chat) == OpenAI:
gpt_output += chunk
cost += float(str(cb).split('$')[1])
true_topics = dataset.loc[idx][truth_column]
accuracies.append(get_accuracy(gpt_output, true_topics))
except Exception as e:
print('Error encountered at index', idx)
print(e)
return accuracies, cost, io_pairs
io_pairs.append([abstract, ', '.join(true_topics), gpt_output])
print('Average Accuracy:', np.mean(accuracies))
print('Total Cost ($):', cost)
return accuracies, cost, io_pairs
</code>
<code>
def get_accuracy(generated_topics, true_topics):
num_correct = 0
for topic in generated_topics.strip().split(', '):
if topic in true_topics:
num_correct += 1
return num_correct / len(true_topics)
</code>
<code>
n = 100
training_data = dataset.sample(n=n, replace=False, random_state=50)
training_data.shape
</code>
<code>
testing_data = dataset.drop(training_data.index)
testing_data['Abstract'] = testing_data['Abstract'].apply(lambda text: BeautifulSoup(text, "html.parser").get_text())
</code>
<code>
results = pd.DataFrame(columns=['Model', 'Abstract', 'Ground Truth', 'Predictions'])
</code>
<code>
# 100 training samples (11/06 version)
chat = ChatOpenAI(
model_name='ft:gpt-3.5-turbo-1106:personal::8SDAGTmv',
openai_api_key = openai_api_key,
request_timeout=120,
max_retries=12
)
accuracies, cost, io_pairs = test(chat, testing_data, truth_column='Filtered EDAM', iterations=25)
for abstract, ground_truth, predictions in io_pairs:
row = {'Model': chat.model_name, 'Abstract': abstract, 'Ground Truth': ground_truth, 'Predictions': predictions}
results = pd.concat([results, pd.DataFrame([row])], ignore_index=True)
</code>
<code>
# 100 training samples (06/13 version)
chat = ChatOpenAI(
model_name='ft:gpt-3.5-turbo-0613:personal::8SD8i1on',
openai_api_key = openai_api_key,
request_timeout=120,
max_retries=12
)
accuracies, cost, io_pairs = test(chat, testing_data, truth_column='Filtered EDAM', iterations=25)
for abstract, ground_truth, predictions in io_pairs:
row = {'Model': chat.model_name, 'Abstract': abstract, 'Ground Truth': ground_truth, 'Predictions': predictions}
results = pd.concat([results, pd.DataFrame([row])], ignore_index=True)
</code>
<code>
# 50 training samples (06/13 version)
chat = ChatOpenAI(
model_name='ft:gpt-3.5-turbo-0613:personal::8SAHvdnS',
openai_api_key = openai_api_key,
request_timeout=120,
max_retries=12
)
accuracies, cost, io_pairs = test(chat, testing_data, truth_column='Filtered EDAM', iterations=25)
for abstract, ground_truth, predictions in io_pairs:
row = {'Model': chat.model_name, 'Abstract': abstract, 'Ground Truth': ground_truth, 'Predictions': predictions}
results = pd.concat([results, pd.DataFrame([row])], ignore_index=True)
</code>
<code>
# Non-fine tuned version. Default dated model
chat = ChatOpenAI(
model_name='gpt-3.5-turbo',
openai_api_key = openai_api_key,
request_timeout=120,
max_retries=12
)
accuracies, cost, io_pairs = test(chat, testing_data, truth_column='Filtered EDAM', iterations=25)
for abstract, ground_truth, predictions in io_pairs:
row = {'Model': chat.model_name, 'Abstract': abstract, 'Ground Truth': ground_truth, 'Predictions': predictions}
results = pd.concat([results, pd.DataFrame([row])], ignore_index=True)
</code>
<code>
# Non fine-tuned GPT 4
chat = ChatOpenAI(
model_name='gpt-4',
openai_api_key = openai_api_key,
request_timeout=120,
max_retries=12
)
accuracies, cost, io_pairs = test(chat, testing_data, truth_column='Filtered EDAM', iterations=25)
for abstract, ground_truth, predictions in io_pairs:
row = {'Model': chat.model_name, 'Abstract': abstract, 'Ground Truth': ground_truth, 'Predictions': predictions}
results = pd.concat([results, pd.DataFrame([row])], ignore_index=True)
</code>
<code>
# import csv
## Convert Predictions column to a set
# results['Predictions'] = results['Predictions'].apply(lambda x: set(map(str.strip, next(csv.reader([x])))))
</code>
<code>
results.to_csv('raw_model_outputs.csv', index=False)
</code>
<code>
def print_dynamically(string, max_line_length=80):
words = string.split()
lines = []
current_line = ""
for word in words:
if len(current_line) + len(word) + 1 <= max_line_length:
current_line += word + " "
else:
lines.append(current_line)
current_line = word + " "
lines.append(current_line)
for line in lines:
print(line)
</code>
<code>
model, abstract, ground_truth, pred = results.sample(n=1).values[0]
print_dynamically(abstract)
print('\nModel:', model)
print('\nGround Truth EDAM Topics:')
print(ground_truth)
print('\nGPT Predicted EDAM Topics:')
print(pred)
</code>
<code>
## Compare old template and new template in terms of hallucinations
halluc_scores = []
for idx, row in results.iterrows():
preds, upd_preds = list(ast.literal_eval(row['Predictions'])), row['New Predictions'].split(', ')
# print(preds)
# print(upd_preds, '\n')
pred_scores = [1 if pred not in full_edam_topics else 0 for pred in preds]
pred_score = sum(pred_scores) / len(pred_scores)
upd_pred_scores = [1 if pred not in full_edam_topics else 0 for pred in upd_preds]
upd_pred_score = sum(upd_pred_scores) / len(upd_pred_scores)
halluc_score = pred_score - upd_pred_score
halluc_scores.append(halluc_score)
</code>
<code>
# Positive means old preds were worse, negative means they were better
plt.hist(halluc_scores, bins='auto', edgecolor='black', alpha=0.7)
plt.title('Hallucination Scores')
plt.xlabel('Hallucination Score')
plt.ylabel('Frequency')
</code>
|
{
"filename": "GPT_1.ipynb",
"repository": "ZubairQazi/NDE-GPT",
"query": "transformed_from_existing",
"size": 117418,
"sha": ""
}
|
# New_eng_academic_research_2.ipynb
Repository: kdj0712/teamKim1
<code>
import pandas as pd
import numpy as np
</code>
<code>
df_Riss_research = pd.read_csv("./csv/Seleniums.eng_academic_research.csv")
df_Riss_research.drop(labels='_id', axis=1, inplace=True)
df_Riss_research['research_subject']
</code>
## 데이터 전처리
### dataframe 내 중복되는 학술정보 제거
<code>
df_Riss_research['research_title'].value_counts()
# 중복되는 research 확인
# 1) 진행성 화골성 근염 -증례 보고- = Myositis Ossificans Progressive -A Case Report-
# 2) 비장적출로 치유된 희귀 비장 질환 치험 = Clinical Experience of Rare Splenic Disease Healed by Splenectomy
# 3) 상급종합병원과 희귀난치성질환 전문병원의 희귀의약품 사용현황
</code>
<code>
df_Riss_research.drop_duplicates(subset="research_title", keep='first', inplace=True)
df_Riss_research['research_title'].value_counts()
</code>
<code>
df_Riss_research['research_title'].value_counts()
# 더이상 중복값 없음을 확인 완료
</code>
<code>
df_Riss_research.reset_index(drop=True, inplace=True)
</code>
### 주제어 존재하는 column만 추출
<code>
drop_index = df_Riss_research[df_Riss_research['research_subject'].str.contains(';')==True].index
</code>
<code>
df_Riss_research_subject = df_Riss_research[df_Riss_research['research_subject'].str.contains(';')==True]
df_Riss_research_subject.reset_index(drop=True, inplace=True)
df_Riss_research_subject
</code>
<code>
condition = "research_language != 'KCI등재후보'"
df_Riss_research_subject01 = df_Riss_research_subject.query(condition)
df_Riss_research_subject01.reset_index(drop=True, inplace=True)
df_Riss_research_subject01
</code>
<code>
type(df_Riss_research_subject01['research_type'][3])
</code>
<code>
int(df_Riss_research_subject01['research_type'][3])
</code>
<code>
for i in range(len(df_Riss_research_subject01['research_type'].index)):
try:
if type(int(df_Riss_research_subject01['research_type'][i])) == int:
condition03 = "research_page != '학술저널'"
df_Riss_research_subject02 = df_Riss_research_subject01.query(condition03)
except:
pass
df_Riss_research_subject02.reset_index(drop=True, inplace=True)
</code>
<code>
df_new = df_Riss_research_subject02[['research_title', 'research_subject']]
df_new.to_csv("eng_research_subject.csv", sep='\t', encoding='utf-8')
</code>
### research_title 영문명만 분리
<code>
import re
def no_korean(text):
patterns = '([가-힣]|[一-龥]|[0-9]|[;])'
text_regex = re.sub(pattern=patterns, repl=' ', string=text)
return text_regex
df_Riss_research_subject['research_subject'] = df_Riss_research_subject['research_subject'].apply(no_korean)
</code>
<code>
df_Riss_research_subject['research_subject']
</code>
<code>
df_new =pd.DataFrame(df_Riss_research_subject['research_subject'])
df_new
</code>
<code>
df_new.to_csv("eng_research_subject.csv", sep='\t', encoding='utf-8')
</code>
<code>
eng_subject = df_Riss_research_subject['research_subject'].tolist()
eng_subject
</code>
### 형태소 분석기
#### 불용어리스트 만들기
<code>
f=open('./csv/eng_academic_research_stopwords.txt')
stopwords=[]
lines = f.readlines()
for line in lines:
line = line.strip()
stopwords.append(line)
f.close()
</code>
<code>
df_Riss_research_subject['research_subject'] = df_Riss_research_subject['research_subject'].str.lower()
</code>
<code>
from sklearn.feature_extraction.text import TfidfVectorizer
tfidfVectorizer = TfidfVectorizer(stop_words=stopwords
, ngram_range=(1,2)
, max_df=0.90
, min_df=1) # stop_words는 vocabulary에서 필요없는 단어를 빼주는 것. ngram_range는 단어를 붙여주는 것으로 2개의 단어가 합성되었을때 의미를 가지고, 떨어져있을때 의미가 상실되는 경우를 포함함.
result_vectors = tfidfVectorizer.fit_transform(eng_subject) # fit & transform은 다른 2가지 임.(fit하면 각 단어의 vocabulary 만들 수 있음.)
result_vectors.toarray()[:2]
</code>
<code>
tfidfVectorizer.vocabulary_
</code>
<code>
from sklearn.decomposition import LatentDirichletAllocation
lda_model = LatentDirichletAllocation(n_components=3, n_jobs=-1) #인스턴스화 #n_components 토픽의 갯수
lda_model.fit(result_vectors) #교육
</code>
<code>
dictionary_list = tfidfVectorizer.get_feature_names_out()
dictionary_list
</code>
<code>
lda_model.components_
</code>
<code>
topics_output = lda_model.transform(result_vectors)
df_topics_score = pd.DataFrame(data=topics_output)
df_topics_score
</code>
<code>
df_topics_score['dominant_topic_number'] = np.argmax(topics_output, axis=1)
df_topics_score['sentences'] = df_Riss_research_subject['research_subject']
df_topics_score
</code>
### topic별 word 추출
<code>
## 상위 단어 추출
## 0 확률 1은 dictionary
topics_list = list()
for topic in lda_model.components_:
df_datas = [topic, dictionary_list]
df_topics = pd.DataFrame(data=df_datas)
df_topics= df_topics.T
df_topics = df_topics.sort_values(0, ascending=False)
# print(df_topics[:3])
topics_text = ' '.join(df_topics[1].values[:3])# 시리즈 형식으로 출력 get values from series / index
print(topics_text)
topics_list.append(topics_text)
topics_list_add = [topics_list, ['Topic0', 'Topic1', 'Topic2']]
df_topics_keywords = pd.DataFrame(topics_list_add)
</code>
<code>
df_topics_keywords
</code>
<code>
import pyLDAvis
import pyLDAvis.lda_model
</code>
<code>
vis = pyLDAvis.lda_model.prepare(lda_model, result_vectors, tfidfVectorizer) # 토픽모델, 교육이 끝난 값(행렬형태), 교육모델
</code>
<code>
pyLDAvis.enable_notebook()
pyLDAvis.display(vis) # PCA - 차원축소
</code>
|
{
"filename": "New_eng_academic_research_2.ipynb",
"repository": "kdj0712/teamKim1",
"query": "transformed_from_existing",
"size": 277407,
"sha": ""
}
|
# analyses_3.SCENIC-V10-V2_1.ipynb
Repository: aertslab/scenicplus
### 1. Create SCENIC+ object
<code>
# Load functions
from scenicplus.scenicplus_class import SCENICPLUS, create_SCENICPLUS_object
from scenicplus.preprocessing.filtering import *
</code>
First we will load the scRNA-seq and the scATAC-seq data. We make sure that names match between them.
<code>
# Load data
## ATAC - cisTopic object
outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/pycisTopic/'
import pickle
infile = open(outDir + 'DPCL_cisTopicObject.pkl', 'rb')
cistopic_obj = pickle.load(infile)
infile.close()
## Precomputed imputed data
import pickle
infile = open(outDir + 'DARs/Imputed_accessibility.pkl', 'rb')
imputed_acc_obj = pickle.load(infile)
infile.close()
## RNA - Create Anndata
from loomxpy.loomxpy import SCopeLoom
from pycisTopic.loom import *
import itertools
import anndata
path_to_loom = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/vsn/grnboost/out/data/scRNA_count_matrix.SINGLE_SAMPLE_SCENIC.loom'
loom = SCopeLoom.read_loom(path_to_loom)
metadata = get_metadata(loom)
# Fix names
metadata.index = metadata.index + '___DPLC'
expr_mat = loom.ex_mtx
expr_mat.index = expr_mat.index + '___DPLC'
rna_anndata = anndata.AnnData(X=expr_mat)
rna_anndata.obs = metadata
</code>
Next we load the motif enrichment results into a dictionary. We can load motif results from the different methods in pycistarget (e.g. cisTarget, DEM) and different region sets (e.g. topics, DARs, MACS bdgdiff peaks). In this tutorial we will use both cisTarget and DEM peaks from topics and DARs.
<code>
## Precomputed imputed data
import pickle
infile = open('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/pycistarget/cluster_V10_V2/menr.pkl', 'rb')
menr = pickle.load(infile)
infile.close()
</code>
Now we can create the SCENIC+ object:
<code>
scplus_obj = create_SCENICPLUS_object(
GEX_anndata = rna_anndata,
cisTopic_obj = cistopic_obj,
imputed_acc_obj = imputed_acc_obj,
menr = menr,
ACC_prefix = 'ACC_',
GEX_prefix = 'GEX_',
bc_transform_func = lambda x: x,
normalize_imputed_acc = False)
</code>
<code>
type(scplus_obj.X_EXP)
</code>
<code>
print(scplus_obj)
</code>
You can also filter low accessible regions and low expressed genes. This recommended to avoid getting false relationships with these regions and genes.
<code>
filter_genes(scplus_obj, min_pct = 0.5)
filter_regions(scplus_obj, min_pct = 0.5)
</code>
<code>
# Save
outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final/'
import pickle
with open(outDir+'scplus_obj.pkl', 'wb') as f:
pickle.dump(scplus_obj, f)
</code>
### GRNBoost
<code>
singularity exec -B /lustre1,/staging,/data,/vsc-hard-mounts,/scratch scenicplus.sif ipython3
</code>
<code>
# For the downstream analyses
outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final/'
import pickle
infile = open(outDir+'scplus_obj.pkl', 'rb')
scplus_obj = pickle.load(infile)
infile.close()
import pickle
infile = open('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_V2_grnboost/region_ranking.pkl', 'rb')
region_ranking = pickle.load(infile)
infile.close()
import pickle
infile = open('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_V2_grnboost/gene_ranking.pkl', 'rb')
gene_ranking = pickle.load(infile)
infile.close()
from scenicplus.wrappers.run_scenicplus import *
run_scenicplus(scplus_obj,
variable = ['ACC_Cell_type'],
species = 'hsapiens',
assembly = 'hg38',
tf_file = '/staging/leuven/stg_00002/lcb/cflerin/resources/allTFs_hg38.txt',
save_path = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final_autoreg/grnboost/',
biomart_host = 'http://oct2016.archive.ensembl.org/',
upstream = [1000, 150000],
downstream = [1000, 150000],
calculate_TF_eGRN_correlation = False,
calculate_DEGs_DARs = True,
export_to_loom_file = True,
export_to_UCSC_file = True,
region_ranking=region_ranking,
gene_ranking=gene_ranking,
tree_structure = ('DPCL', 'SCENIC+', 'grnboost'),
path_bedToBigBed = '/data/leuven/software/biomed/haswell_centos7/2018a/software/Kent_tools/20190730-linux.x86_64/bin/',
n_cpu = 20,
_temp_dir = '/scratch/leuven/313/vsc31305/ray_spill'
)
</code>
<code>
# For the downstream analyses
outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final_autoreg/grnboost/'
import dill
infile = open(outDir+'scplus_obj.pkl', 'rb')
scplus_obj = dill.load(infile)
infile.close()
</code>
<code>
import pandas as pd
def format_egrns_time(scplus_obj,
eregulons_key: str = 'eRegulons',
TF2G_key: str = 'TF2G_adj',
key_added: str = 'eRegulon_metadata'):
"""
A function to format eRegulons to a pandas dataframe
"""
egrn_list = scplus_obj.uns[eregulons_key]
TF = [egrn_list[x].transcription_factor for x in range(len(egrn_list))]
is_extended = [str(egrn_list[x].is_extended)
for x in range(len(egrn_list))]
r2g_data = [pd.DataFrame.from_records(egrn_list[x].regions2genes, columns=[
'Region', 'Gene', 'R2G_importance', 'R2G_rho', 'R2G_importance_x_rho', 'R2G_importance_x_abs_rho']) for x in range(len(egrn_list))]
egrn_name = [TF[x] + '_extended' if is_extended[x] ==
'True' else TF[x] for x in range(len(egrn_list))]
egrn_name = [egrn_name[x] + '_+' if 'positive tf2g' in egrn_list[x]
.context else egrn_name[x] + '_-' for x in range(len(egrn_list))]
egrn_name = [egrn_name[x] + '_+' if 'positive r2g' in egrn_list[x]
.context else egrn_name[x] + '_-' for x in range(len(egrn_list))]
region_signature_name = [
egrn_name[x] + '_(' + str(len(set(r2g_data[x].Region))) + 'r)' for x in range(len(egrn_list))]
gene_signature_name = [
egrn_name[x] + '_(' + str(len(set(r2g_data[x].Gene))) + 'g)' for x in range(len(egrn_list))]
for x in range(len(egrn_list)):
r2g_data[x].insert(0, "TF", TF[x])
r2g_data[x].insert(1, "is_extended", is_extended[x])
r2g_data[x].insert(0, "Gene_signature_name", gene_signature_name[x])
r2g_data[x].insert(0, "Region_signature_name",
region_signature_name[x])
tf2g_data = scplus_obj.uns[TF2G_key].copy()
tf2g_data.columns = ['TF', 'Gene', 'TF2G_importance', 'TF2G_regulation',
'TF2G_rho', 'TF2G_importance_x_abs_rho', 'TF2G_importance_x_rho']
egrn_metadata = pd.concat([pd.merge(r2g_data[x], tf2g_data[tf2g_data.TF == r2g_data[x].TF[0]], on=[
'TF', 'Gene']) for x in range(len(egrn_list)) if tf2g_data[tf2g_data.TF == r2g_data[x].TF[0]].shape[0] != 0 and r2g_data[x].shape[0] != 0])
scplus_obj.uns[key_added] = egrn_metadata
</code>
<code>
format_egrns_time(scplus_obj,
eregulons_key = 'eRegulons',
TF2G_key = 'TF2G_adj',
key_added = 'eRegulon_metadata')
</code>
<code>
scplus_obj.uns['region_to_gene'].to_csv('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_V2_grnboost_2/region_to_gene.tsv', sep='\t')
</code>
<code>
scplus_obj.uns['region_to_gene'][scplus_obj.uns['region_to_gene']['rho'] >0].to_csv('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_V2_grnboost_2/region_to_gene_pos.tsv', sep='\t')
</code>
<code>
select = list(set(scplus_obj.uns['eRegulon_metadata']['Region']))
</code>
<code>
scplus_obj.uns['region_to_gene']
</code>
<code>
scplus_obj.uns['region_to_gene'][scplus_obj.uns['region_to_gene']['region'].isin(select)]
</code>
<code>
scplus_obj.uns['region_to_gene'][scplus_obj.uns['region_to_gene']['region'].isin(select)].to_csv('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_V2_grnboost_2/region_to_gene_in_eGRN.tsv', sep='\t')
</code>
<code>
import pandas as pd
hic_data = pd.read_csv('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/formatted/HepG2_ENCFF020DPP_5Kb_SCALE.txt', sep='\t')
hic_data.columns = ['Chromosome', 'Start', 'End', 'target', 'importance', 'TSS', 'Strand', 'Distance', 'region']
hic_data['rho'] = hic_data['importance']
hic_data = hic_data[['target', 'region', 'importance', 'rho', 'Distance']]
hic_data = hic_data[hic_data['region'].isin(scplus_obj.region_names)]
hic_data
</code>
<code>
import pandas as pd
import numpy as np
import ray
import logging
import time
import sys
import os
import subprocess
import pyranges as pr
from sklearn.ensemble import GradientBoostingRegressor, RandomForestRegressor, ExtraTreesRegressor
from scipy.stats import pearsonr, spearmanr
from tqdm import tqdm
from matplotlib import cm
from matplotlib.colors import Normalize
from typing import List
from scenicplus.utils import extend_pyranges, extend_pyranges_with_limits, reduce_pyranges_with_limits_b
from scenicplus.utils import calculate_distance_with_limits_join, reduce_pyranges_b, calculate_distance_join
from scenicplus.utils import coord_to_region_names, region_names_to_coordinates, ASM_SYNONYMS, Groupby, flatten_list
from scenicplus.scenicplus_class import SCENICPLUS
from scenicplus.enhancer_to_gene import INTERACT_AS
def export_to_UCSC_interact_hic(SCENICPLUS_obj: SCENICPLUS,
species: str,
outfile: str,
region_to_gene_key: str =' region_to_gene',
pbm_host:str = 'http://www.ensembl.org',
bigbed_outfile:str = None,
path_bedToBigBed: str= None,
assembly: str = None,
ucsc_track_name: str = 'region_to_gene',
ucsc_description: str = 'interaction file for region to gene',
cmap_neg: str = 'Reds',
cmap_pos: str = 'Greens',
key_for_color: str = 'importance',
vmin: int = 0,
vmax: int = 1,
scale_by_gene: bool = True,
subset_for_eRegulons_regions: bool = True,
eRegulons_key: str = 'eRegulons') -> pd.DataFrame:
# Create logger
level = logging.INFO
format = '%(asctime)s %(name)-12s %(levelname)-8s %(message)s'
handlers = [logging.StreamHandler(stream=sys.stdout)]
logging.basicConfig(level=level, format=format, handlers=handlers)
log = logging.getLogger('R2G')
if region_to_gene_key not in SCENICPLUS_obj.uns.keys():
raise Exception(
f'key {region_to_gene_key} not found in SCENICPLUS_obj.uns, first calculate region to gene relationships using function: "calculate_regions_to_genes_relationships"')
region_to_gene_df = SCENICPLUS_obj.uns[region_to_gene_key].copy()
if subset_for_eRegulons_regions:
if eRegulons_key not in SCENICPLUS_obj.uns.keys():
raise ValueError(
f'key {eRegulons_key} not found in SCENICPLUS_obj.uns.keys()')
eRegulon_regions = list(set(flatten_list(
[ereg.target_regions for ereg in SCENICPLUS_obj.uns[eRegulons_key]])))
region_to_gene_df.index = region_to_gene_df['region']
region_to_gene_df = region_to_gene_df.loc[eRegulon_regions].reset_index(
drop=True)
# Rename columns to be in line with biomart annotation
region_to_gene_df.rename(columns={'target': 'Gene'}, inplace=True)
# Get TSS annotation (end-point for links)
log.info('Downloading gene annotation from biomart, using dataset: {}'.format(
species+'_gene_ensembl'))
import pybiomart as pbm
dataset = pbm.Dataset(name=species+'_gene_ensembl', host=pbm_host)
annot = dataset.query(attributes=['chromosome_name', 'start_position', 'end_position',
'strand', 'external_gene_name', 'transcription_start_site', 'transcript_biotype'])
annot.columns = ['Chromosome', 'Start', 'End', 'Strand',
'Gene', 'Transcription_Start_Site', 'Transcript_type']
annot['Chromosome'] = 'chr' + \
annot['Chromosome'].astype(str)
annot = annot[annot.Transcript_type == 'protein_coding']
annot.Strand[annot.Strand == 1] = '+'
annot.Strand[annot.Strand == -1] = '-'
log.info('Formatting data ...')
# get gene to tss mapping, take the one equal to the gene start/end location if possible otherwise take the first one
annot['TSSeqStartEnd'] = np.logical_or(
annot['Transcription_Start_Site'] == annot['Start'], annot['Transcription_Start_Site'] == annot['End'])
gene_to_tss = annot[['Gene', 'Transcription_Start_Site']].groupby(
'Gene').agg(lambda x: list(map(str, x)))
startEndEq = annot[['Gene', 'TSSeqStartEnd']
].groupby('Gene').agg(lambda x: list(x))
gene_to_tss['Transcription_Start_Site'] = [np.array(tss[0])[eq[0]][0] if sum(
eq[0]) >= 1 else tss[0][0] for eq, tss in zip(startEndEq.values, gene_to_tss.values)]
gene_to_tss.columns = ['TSS_Gene']
# get gene to strand mapping
gene_to_strand = annot[['Gene', 'Strand']].groupby(
'Gene').agg(lambda x: list(map(str, x))[0])
# get gene to chromosome mapping (should be the same as the regions mapped to the gene)
gene_to_chrom = annot[['Gene', 'Chromosome']].groupby(
'Gene').agg(lambda x: list(map(str, x))[0])
# add TSS for each gene to region_to_gene_df
region_to_gene_df = region_to_gene_df.join(gene_to_tss, on='Gene')
# add strand for each gene to region_to_gene_df
region_to_gene_df = region_to_gene_df.join(gene_to_strand, on='Gene')
# add chromosome for each gene to region_to_gene_df
region_to_gene_df = region_to_gene_df.join(gene_to_chrom, on='Gene')
# get chrom, chromStart, chromEnd
region_to_gene_df.dropna(axis=0, how='any', inplace=True)
arr = region_names_to_coordinates(region_to_gene_df['region']).to_numpy()
chrom, chromStart, chromEnd = np.split(arr, 3, 1)
chrom = chrom[:, 0]
chromStart = chromStart[:, 0]
chromEnd = chromEnd[:, 0]
# get source chrom, chromStart, chromEnd (i.e. middle of regions)
sourceChrom = chrom
sourceStart = np.array(
list(map(int, chromStart + (chromEnd - chromStart)/2 - 1)))
sourceEnd = np.array(
list(map(int, chromStart + (chromEnd - chromStart)/2)))
# get target chrom, chromStart, chromEnd (i.e. TSS)
targetChrom = region_to_gene_df['Chromosome']
targetStart = region_to_gene_df['TSS_Gene'].values
targetEnd = list(map(str, np.array(list(map(int, targetStart))) + np.array(
[1 if strand == '+' else -1 for strand in region_to_gene_df['Strand'].values])))
# get color
norm = Normalize(vmin=vmin, vmax=vmax)
if scale_by_gene:
grouper = Groupby(
region_to_gene_df.loc[:, 'Gene'].to_numpy())
scores = region_to_gene_df.loc[:, key_for_color].to_numpy()
mapper = cm.ScalarMappable(norm=norm, cmap=cmap_pos)
def _value_to_color(scores):
S = (scores - scores.min()) / (scores.max() - scores.min())
return [','.join([str(x) for x in mapper.to_rgba(s, bytes=True)][0:3]) for s in S]
colors_pos = np.zeros(len(scores), dtype='object')
for idx in grouper.indices:
colors_pos[idx] = _value_to_color(scores[idx])
def _value_to_color(scores):
S = (scores - scores.min()) / (scores.max() - scores.min())
return [','.join([str(x) for x in mapper.to_rgba(s, bytes=True)][0:3]) for s in S]
colors_neg = np.zeros(len(scores), dtype='object')
for idx in grouper.indices:
colors_neg[idx] = _value_to_color(scores[idx])
else:
scores = region_to_gene_df.loc[:, key_for_color].to_numpy()
mapper = cm.ScalarMappable(norm=norm, cmap=cmap_pos)
colors_pos = [
','.join([str(x) for x in mapper.to_rgba(s, bytes=True)][0:3]) for s in scores]
region_to_gene_df.loc[:, 'color'] = colors_pos
region_to_gene_df['color'] = region_to_gene_df['color'].fillna('55,55,55')
# get name for regions (add incremental number to gene in range of regions linked to gene)
counter = 1
previous_gene = region_to_gene_df['Gene'].values[0]
names = []
for gene in region_to_gene_df['Gene'].values:
if gene != previous_gene:
counter = 1
else:
counter += 1
names.append(gene + '_' + str(counter))
previous_gene = gene
# format final interact dataframe
df_interact = pd.DataFrame(
data={
'chrom': chrom,
'chromStart': chromStart,
'chromEnd': chromEnd,
'name': names,
'score': (1000*(region_to_gene_df['importance'].values - np.min(region_to_gene_df['importance'].values))/np.ptp(region_to_gene_df['importance'].values)).astype(int) ,
'value': region_to_gene_df['importance'].values,
'exp': np.repeat('.', len(region_to_gene_df)),
'color': region_to_gene_df['color'].values,
'sourceChrom': sourceChrom,
'sourceStart': sourceStart,
'sourceEnd': sourceEnd,
'sourceName': names,
'sourceStrand': np.repeat('.', len(region_to_gene_df)),
'targetChrom': targetChrom,
'targetStart': targetStart,
'targetEnd': targetEnd,
'targetName': region_to_gene_df['Gene'].values,
'targetStrand': region_to_gene_df['Strand'].values
}
)
# sort dataframe
df_interact = df_interact.sort_values(by=['chrom', 'chromStart'])
# Write interact file
log.info('Writing data to: {}'.format(outfile))
with open(outfile, 'w') as f:
f.write('track type=interact name="{}" description="{}" useScore=0 maxHeightPixels=200:100:50 visibility=full\n'.format(
ucsc_track_name, ucsc_description))
df_interact.to_csv(f, header=False, index=False, sep='\t')
# write bigInteract file
if bigbed_outfile != None:
log.info('Writing data to: {}'.format(bigbed_outfile))
outfolder = bigbed_outfile.rsplit('/', 1)[0]
# write bed file without header to tmp file
df_interact.to_csv(os.path.join(
outfolder, 'interact.bed.tmp'), header=False, index=False, sep='\t')
# check if auto sql definition for interaction file exists in outfolder, otherwise create it
if not os.path.exists(os.path.join(outfolder, 'interact.as')):
with open(os.path.join(outfolder, 'interact.as'), 'w') as f:
f.write(INTERACT_AS)
# convert interact.bed.tmp to bigBed format
# bedToBigBed -as=interact.as -type=bed5+13 region_to_gene_no_head.interact https://genome.ucsc.edu/goldenPath/help/hg38.chrom.sizes region_to_gene.inter.bb
cmds = [
os.path.join(path_bedToBigBed, 'bedToBigBed'),
'-as={}'.format(os.path.join(os.path.join(outfolder, 'interact.as'))),
'-type=bed5+13',
os.path.join(outfolder, 'interact.bed.tmp'),
'https://hgdownload.cse.ucsc.edu/goldenpath/' + assembly + '/bigZips/' + assembly + '.chrom.sizes',
bigbed_outfile
]
p = subprocess.Popen(cmds, stdout=subprocess.PIPE,
stderr=subprocess.PIPE)
stdout, stderr = p.communicate()
if p.returncode:
raise ValueError(
"cmds: %s\nstderr:%s\nstdout:%s" % (
" ".join(cmds), stderr, stdout)
)
return df_interact
</code>
# Generate Hi-C files
<code>
# For the downstream analyses
outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final_autoreg/grnboost/'
import dill
infile = open(outDir+'scplus_obj.pkl', 'rb')
scplus_obj = dill.load(infile)
infile.close()
</code>
<code>
r2g = scplus_obj.uns['region_to_gene'].copy()
r2g = r2g[r2g.rho > 0.03]
r2g
</code>
<code>
scplus_obj.uns['region_to_gene'].to_csv('/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final_autoreg/grnboost/region_to_gene_pos.tsv', sep='\t')
</code>
<code>
import os
from os import listdir
from os.path import isfile, join
import pandas as pd
cell_lines=['GM12878', 'HepG2', 'IMR90', 'HCT116', 'K562']
save_path = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/bb_files_final/'
species = 'hsapiens'
assembly = 'hg38'
path_bedToBigBed = '/data/leuven/software/biomed/haswell_centos7/2018a/software/Kent_tools/20190730-linux.x86_64/bin/'
biomart_host = 'http://oct2016.archive.ensembl.org/'
path='/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/formatted/'
for cell_line in cell_lines:
files = [f for f in listdir(path) if isfile(join(path, f))]
file = [f for f in files if cell_line in f]
hic_data = pd.read_csv(path+file[0], sep='\t')
hic_data.columns = ['Chromosome', 'Start', 'End', 'target', 'importance', 'TSS', 'Strand', 'Distance', 'region']
hic_data['rho'] = hic_data['importance']
hic_data = hic_data[['target', 'region', 'importance', 'rho', 'Distance']]
hic_data = hic_data[hic_data['region'].isin(scplus_obj.region_names)]
scplus_obj.uns['region_to_gene'] = hic_data
r2g_data = export_to_UCSC_interact_hic(scplus_obj,
species,
os.path.join(save_path,cell_line+'.hic.all.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,cell_line+'.hic.all.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description=cell_line+' HiC links',
cmap_neg='Reds',
cmap_pos='Greys',
key_for_color='importance',
scale_by_gene=True,
subset_for_eRegulons_regions=False,
eRegulons_key='eRegulons')
</code>
<code>
import os
from os import listdir
from os.path import isfile, join
import pandas as pd
cell_lines=['CellOracle', 'FigR', 'GRaNIE', 'Scenicplus-importance', 'Scenicplus-rho']
color_dict={'CellOracle':'Greens', 'FigR':'Purples', 'GRaNIE':'Oranges', 'Scenicplus-importance':'Blues', 'Scenicplus-rho':'Reds'}
save_path = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/bb_files_final/'
species = 'hsapiens'
assembly = 'hg38'
path_bedToBigBed = '/data/leuven/software/biomed/haswell_centos7/2018a/software/Kent_tools/20190730-linux.x86_64/bin/'
biomart_host = 'http://oct2016.archive.ensembl.org/'
path='/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/formatted/'
for cell_line in cell_lines:
files = [f for f in listdir(path) if isfile(join(path, f))]
file = [f for f in files if cell_line in f]
hic_data = pd.read_csv(path+file[0], sep='\t')
hic_data.columns = ['Chromosome', 'Start', 'End', 'target', 'importance', 'TSS', 'Strand', 'Distance', 'region']
hic_data['rho'] = hic_data['importance']
hic_data = hic_data[['target', 'region', 'importance', 'rho', 'Distance']]
hic_data = hic_data[hic_data['region'].isin(scplus_obj.region_names)]
scplus_obj.uns['region_to_gene'] = hic_data
r2g_data = export_to_UCSC_interact_hic(scplus_obj,
species,
os.path.join(save_path,cell_line+'.links.all.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,cell_line+'.links.all.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description='SCENIC+ region to gene links',
cmap_neg='Reds',
cmap_pos=color_dict[cell_line],
key_for_color='importance',
scale_by_gene=True,
subset_for_eRegulons_regions=False,
eRegulons_key='eRegulons')
</code>
<code>
import os
from os import listdir
from os.path import isfile, join
import pandas as pd
cell_lines=['Scenicplus-importance_links_all', 'Scenicplus-rho_links_all']
color_dict={'Scenicplus-importance_links_all':'Blues', 'Scenicplus-rho_links_all':'Reds'}
save_path = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/bb_files_final/'
species = 'hsapiens'
assembly = 'hg38'
path_bedToBigBed = '/data/leuven/software/biomed/haswell_centos7/2018a/software/Kent_tools/20190730-linux.x86_64/bin/'
biomart_host = 'http://oct2016.archive.ensembl.org/'
path='/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/data/HiC/formatted/'
for cell_line in cell_lines:
files = [f for f in listdir(path) if isfile(join(path, f))]
file = [f for f in files if cell_line in f]
hic_data = pd.read_csv(path+file[0], sep='\t')
hic_data.columns = ['Chromosome', 'Start', 'End', 'target', 'importance', 'TSS', 'Strand', 'Distance', 'region']
hic_data['rho'] = hic_data['importance']
hic_data = hic_data[['target', 'region', 'importance', 'rho', 'Distance']]
hic_data = hic_data[hic_data['region'].isin(scplus_obj.region_names)]
scplus_obj.uns['region_to_gene'] = hic_data
r2g_data = export_to_UCSC_interact_hic(scplus_obj,
species,
os.path.join(save_path,cell_line+'.links.all.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,cell_line+'.links.all.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description='SCENIC+ region to gene links',
cmap_neg='Reds',
cmap_pos=color_dict[cell_line],
key_for_color='importance',
scale_by_gene=True,
subset_for_eRegulons_regions=False,
eRegulons_key='eRegulons')
</code>
<code>
#from scenicplus.enhancer_to_gene import export_to_UCSC_interact
</code>
<code>
hic_data
</code>
<code>
eRegulon_regions = list(set(flatten_list([ereg.target_regions for ereg in scplus_obj.uns['eRegulons']])))
hic_data = hic_data[hic_data['region'].isin(eRegulon_regions)]
scplus_obj.uns['region_to_gene'] = hic_data
r2g_data = export_to_UCSC_interact_hic(scplus_obj,
species,
os.path.join(save_path,'HepG2.hic.eGRN.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,'HepG2.hic.eGRN.notscaled.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description='SCENIC+ region to gene links',
cmap_neg='Reds',
cmap_pos='Greens',
key_for_color='importance',
scale_by_gene=False,
subset_for_eRegulons_regions=False,
eRegulons_key='eRegulons')
</code>
<code>
# For the downstream analyses
#outDir = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final/'
outDir = '/data/users/cbravo/DPCL/scenicplus_final_autoreg/'
import pickle
infile = open(outDir+'scplus_obj.pkl', 'rb')
scplus_obj = pickle.load(infile)
infile.close()
import pickle
infile = open('/data/users/cbravo/DPCL/scenicplus_final_autoreg/genie3/region_ranking.pkl', 'rb')
region_ranking = pickle.load(infile)
infile.close()
import pickle
infile = open('/data/users/cbravo/DPCL/scenicplus_final_autoreg/genie3/gene_ranking.pkl', 'rb')
gene_ranking = pickle.load(infile)
infile.close()
from scenicplus.wrappers.run_scenicplus import *
run_scenicplus_genie3(scplus_obj,
variable = ['ACC_Cell_type'],
species = 'hsapiens',
assembly = 'hg38',
tf_file = '/data/users/cbravo/resources/allTFs_hg38.txt',
save_path = '/data/users/cbravo/DPCL/scenicplus_final_autoreg/genie3/',
biomart_host = 'http://oct2016.archive.ensembl.org/',
upstream = [1000, 150000],
downstream = [1000, 150000],
calculate_TF_eGRN_correlation = False,
calculate_DEGs_DARs = True,
export_to_loom_file = True,
export_to_UCSC_file = True,
region_ranking=region_ranking,
gene_ranking=gene_ranking,
tree_structure = ('DPCL', 'SCENIC+', 'genie3'),
path_bedToBigBed = '/media/data/users/cbravo/software/KENT/',
n_cpu = 20,
_temp_dir = '/media/data/users/cbravo/ray_spill'
)
from scenicplus.wrappers.run_scenicplus import *
run_scenicplus_genie3(scplus_obj,
variable = ['ACC_Cell_type'],
species = 'hsapiens',
assembly = 'hg38',
tf_file = '/staging/leuven/stg_00002/lcb/cflerin/resources/allTFs_hg38.txt',
save_path = '/staging/leuven/stg_00002/lcb/cbravo/Multiomics_pipeline/analysis/DPCL/scenicplus_final_autoreg/genie3/',
biomart_host = 'http://oct2016.archive.ensembl.org/',
upstream = [1000, 150000],
downstream = [1000, 150000],
calculate_TF_eGRN_correlation = False,
calculate_DEGs_DARs = True,
export_to_loom_file = True,
export_to_UCSC_file = True,
tree_structure = ('DPCL', 'SCENIC+', 'genie3'),
path_bedToBigBed = '/data/leuven/software/biomed/haswell_centos7/2018a/software/Kent_tools/20190730-linux.x86_64/bin/',
n_cpu = 14,
_temp_dir = '/scratch/leuven/313/vsc31305/ray_spill'
)
</code>
<code>
from scenicplus.scenicplus_class import SCENICPLUS, create_SCENICPLUS_object
from scenicplus.preprocessing.filtering import *
from scenicplus.cistromes import *
from scenicplus.enhancer_to_gene import get_search_space, calculate_regions_to_genes_relationships, RF_KWARGS
from scenicplus.enhancer_to_gene import export_to_UCSC_interact
from scenicplus.utils import format_egrns, export_eRegulons
from scenicplus.eregulon_enrichment import *
from scenicplus.TF_to_gene import *
from scenicplus.grn_builder.gsea_approach import build_grn
from scenicplus.dimensionality_reduction import *
from scenicplus.RSS import *
from scenicplus.diff_features import *
from scenicplus.loom import *
from typing import Dict, List, Mapping, Optional, Sequence
import os
import dill
import time
def run_scenicplus_genie3(scplus_obj: 'SCENICPLUS',
variable: List[str],
species: str,
assembly: str,
tf_file: str,
save_path: str,
biomart_host: Optional[str] = 'http://www.ensembl.org',
upstream: Optional[List] = [1000, 150000],
downstream: Optional[List] = [1000, 150000],
region_ranking: Optional['CisTopicImputedFeatures'] = None,
gene_ranking: Optional['CisTopicImputedFeatures'] = None,
calculate_TF_eGRN_correlation: Optional[bool] = True,
calculate_DEGs_DARs: Optional[bool] = True,
export_to_loom_file: Optional[bool] = True,
export_to_UCSC_file: Optional[bool] = True,
tree_structure: Sequence[str] = (),
path_bedToBigBed: Optional[str] = None,
n_cpu: Optional[int] = 1,
_temp_dir: Optional[str] = '/scratch/leuven/313/vsc31305/ray_spill'
):
"""
Wrapper to run SCENIC+
Parameters
---------
scplus_obj: `class::SCENICPLUS`
A SCENICPLUS object.
variables: List[str]
Variables to use for RSS, TF-eGRN correlation and markers.
species: str
Species from which data comes from. Possible values: 'hsapiens', 'mmusculus', 'dmelanogaster'
assembly: str
Genome assembly to which the data was mapped. Possible values: 'hg38'
tf_file: str
Path to file containing genes that are TFs
save_path: str
Folder in which results will be saved
biomart_host: str, optional
Path to biomart host. Make sure that the host matches your genome assembly
upstream: str, optional
Upstream space to use for region to gene relationships
downstream: str, optional
Upstream space to use for region to gene relationships
region_ranking: `class::CisTopicImputedFeatures`, optional
Precomputed region ranking
gene_ranking: `class::CisTopicImputedFeatures`, optional
Precomputed gene ranking
calculate_TF_eGRN_correlation: bool, optional
Whether to calculate the TF-eGRN correlation based on the variables
calculate_DEGs_DARs: bool, optional
Whether to calculate DARs/DEGs based on the variables
export_to_loom_file: bool, optional
Whether to export data to loom files (gene based/region based)
export_to_UCSC_file: bool, optional
Whether to export region-to-gene links and eregulons to bed files
tree_structure: sequence, optional
Tree structure for loom files
path_bedToBigBed: str, optional
Path to convert bed files to big bed when exporting to UCSC (required if files are meant to be
used in a hub)
n_cpu: int, optional
Number of cores to use
_temp_dir: str, optional
Temporary directory for ray
"""
# Create logger
level = logging.INFO
log_format = '%(asctime)s %(name)-12s %(levelname)-8s %(message)s'
handlers = [logging.StreamHandler(stream=sys.stdout)]
logging.basicConfig(level=level, format=log_format, handlers=handlers)
log = logging.getLogger('SCENIC+_wrapper')
start_time = time.time()
check_folder = os.path.isdir(save_path)
if not check_folder:
os.makedirs(save_path)
log.info("Created folder : "+ save_path)
else:
log.info(save_path + " folder already exists.")
if 'Cistromes' not in scplus_obj.uns.keys():
log.info('Merging cistromes')
merge_cistromes(scplus_obj)
if 'search_space' not in scplus_obj.uns.keys():
log.info('Getting search space')
get_search_space(scplus_obj,
biomart_host = biomart_host,
species = species,
assembly = assembly,
upstream = upstream,
downstream = downstream)
if 'region_to_gene' not in scplus_obj.uns.keys():
log.info('Inferring region to gene relationships')
calculate_regions_to_genes_relationships(scplus_obj,
ray_n_cpu = n_cpu,
_temp_dir = _temp_dir,
importance_scoring_method = 'RF',
importance_scoring_kwargs = RF_KWARGS)
if 'TF2G_adj' not in scplus_obj.uns.keys():
log.info('Inferring TF to gene relationships')
calculate_TFs_to_genes_relationships(scplus_obj,
tf_file = tf_file,
ray_n_cpu = n_cpu,
method = 'GBM',
_temp_dir = _temp_dir,
key= 'TF2G_adj')
if 'eRegulons' not in scplus_obj.uns.keys():
log.info('Build eGRN')
build_grn(scplus_obj,
min_target_genes = 10,
adj_pval_thr = 1,
min_regions_per_gene = 0,
quantiles = (0.85, 0.90, 0.95),
top_n_regionTogenes_per_gene = (5, 10, 15),
top_n_regionTogenes_per_region = (),
binarize_using_basc = True,
rho_dichotomize_tf2g = True,
rho_dichotomize_r2g = True,
rho_dichotomize_eregulon = True,
rho_threshold = 0.05,
keep_extended_motif_annot = True,
merge_eRegulons = True,
order_regions_to_genes_by = 'importance',
order_TFs_to_genes_by = 'importance',
key_added = 'eRegulons',
cistromes_key = 'Unfiltered',
disable_tqdm = True,
ray_n_cpu = n_cpu,
_temp_dir = _temp_dir)
if 'eRegulon_metadata' not in scplus_obj.uns.keys():
log.info('Formatting eGRNs')
format_egrns(scplus_obj,
eregulons_key = 'eRegulons',
TF2G_key = 'TF2G_adj',
key_added = 'eRegulon_metadata')
if 'eRegulon_signatures' not in scplus_obj.uns.keys():
log.info('Converting eGRNs to signatures')
get_eRegulons_as_signatures(scplus_obj,
eRegulon_metadata_key='eRegulon_metadata',
key_added='eRegulon_signatures')
#if 'eRegulon_AUC' not in scplus_obj.uns.keys():
log.info('Calculating eGRNs AUC')
if region_ranking is None:
log.info('Calculating region ranking')
region_ranking = make_rankings(scplus_obj, target='region')
with open(os.path.join(save_path,'region_ranking.pkl'), 'wb') as f:
dill.dump(region_ranking, f)
log.info('Calculating eGRNs region based AUC')
score_eRegulons(scplus_obj,
ranking = region_ranking,
eRegulon_signatures_key = 'eRegulon_signatures',
key_added = 'eRegulon_AUC',
enrichment_type= 'region',
auc_threshold = 0.05,
normalize = False,
n_cpu = n_cpu)
if gene_ranking is None:
log.info('Calculating gene ranking')
gene_ranking = make_rankings(scplus_obj, target='gene')
with open(os.path.join(save_path,'gene_ranking.pkl'), 'wb') as f:
dill.dump(gene_ranking, f)
log.info('Calculating eGRNs gene based AUC')
score_eRegulons(scplus_obj,
gene_ranking,
eRegulon_signatures_key = 'eRegulon_signatures',
key_added = 'eRegulon_AUC',
enrichment_type = 'gene',
auc_threshold = 0.05,
normalize= False,
n_cpu = n_cpu)
if calculate_TF_eGRN_correlation is True:
log.info('Calculating TF-eGRNs AUC correlation')
for var in variable:
generate_pseudobulks(scplus_obj,
variable = var,
auc_key = 'eRegulon_AUC',
signature_key = 'Gene_based',
nr_cells = 5,
nr_pseudobulks = 100,
seed=555)
generate_pseudobulks(scplus_obj,
variable = var,
auc_key = 'eRegulon_AUC',
signature_key = 'Region_based',
nr_cells = 5,
nr_pseudobulks = 100,
seed=555)
TF_cistrome_correlation(scplus_obj,
variable = var,
auc_key = 'eRegulon_AUC',
signature_key = 'Gene_based',
out_key = var+'_eGRN_gene_based')
TF_cistrome_correlation(scplus_obj,
variable = var,
auc_key = 'eRegulon_AUC',
signature_key = 'Region_based',
out_key = var+'_eGRN_region_based')
#if 'eRegulon_AUC_thresholds' not in scplus_obj.uns.keys():
log.info('Binarizing eGRNs AUC')
binarize_AUC(scplus_obj,
auc_key='eRegulon_AUC',
out_key='eRegulon_AUC_thresholds',
signature_keys=['Gene_based', 'Region_based'],
n_cpu=n_cpu)
#if 'eRegulons_UMAP' not in scplus_obj.dr_cell.keys():
log.info('Making eGRNs AUC UMAP')
run_eRegulons_umap(scplus_obj,
scale=True, signature_keys=['Gene_based', 'Region_based'])
#if 'eRegulons_tSNE' not in scplus_obj.dr_cell.keys():
log.info('Making eGRNs AUC tSNE')
run_eRegulons_tsne(scplus_obj,
scale=True, signature_keys=['Gene_based', 'Region_based'])
#if 'RSS' not in scplus_obj.uns.keys():
log.info('Calculating eRSS')
for var in variable:
regulon_specificity_scores(scplus_obj,
var,
signature_keys=['Gene_based'],
out_key_suffix='_gene_based',
scale=False)
regulon_specificity_scores(scplus_obj,
var,
signature_keys=['Region_based'],
out_key_suffix='_region_based',
scale=False)
if calculate_DEGs_DARs is True:
log.info('Calculating DEGs/DARs')
for var in variable:
get_differential_features(scplus_obj, var, use_hvg = True, contrast_type = ['DEGs', 'DARs'])
if export_to_loom_file is True:
log.info('Exporting to loom file')
export_to_loom(scplus_obj,
signature_key = 'Gene_based',
tree_structure = tree_structure,
title = 'Gene based eGRN',
nomenclature = assembly,
out_fname=os.path.join(save_path,'SCENIC+_gene_based.loom'))
export_to_loom(scplus_obj,
signature_key = 'Region_based',
tree_structure = tree_structure,
title = 'Region based eGRN',
nomenclature = assembly,
out_fname=os.path.join(save_path,'SCENIC+_region_based.loom'))
if export_to_UCSC_file is True:
log.info('Exporting to UCSC')
r2g_data = export_to_UCSC_interact(scplus_obj,
species,
os.path.join(save_path,'r2g.rho.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,'r2g.rho.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description='SCENIC+ region to gene links',
cmap_neg='Reds',
cmap_pos='Greens',
key_for_color='rho',
scale_by_gene=False,
subset_for_eRegulons_regions=True,
eRegulons_key='eRegulons')
r2g_data = export_to_UCSC_interact(scplus_obj,
species,
os.path.join(save_path,'r2g.importance.bed'),
path_bedToBigBed=path_bedToBigBed,
bigbed_outfile=os.path.join(save_path,'r2g.importance.bb'),
region_to_gene_key='region_to_gene',
pbm_host=biomart_host,
assembly=assembly,
ucsc_track_name='R2G',
ucsc_description='SCENIC+ region to gene links',
cmap_neg='Reds',
cmap_pos='Greens',
key_for_color='importance',
scale_by_gene=True,
subset_for_eRegulons_regions=True,
eRegulons_key='eRegulons')
regions = export_eRegulons(scplus_obj,
os.path.join(save_path,'eRegulons.bed'),
assembly,
bigbed_outfile = os.path.join(save_path,'eRegulons.bb'),
eRegulon_metadata_key = 'eRegulon_metadata',
eRegulon_signature_key = 'eRegulon_signatures',
path_bedToBigBed=path_bedToBigBed)
log.info('Saving object')
with open(os.path.join(save_path,'scplus_obj_genie3.pkl'), 'wb') as f:
dill.dump(scplus_obj, f)
log.info('Finished! Took {} minutes'.format((time.time() - start_time)/60))
</code>
|
{
"filename": "analyses_3.SCENIC-V10-V2_1.ipynb",
"repository": "aertslab/scenicplus",
"query": "transformed_from_existing",
"size": 123784,
"sha": ""
}
|
# generate_chapter_dataset_1.ipynb
Repository: AFF-Learntelligence/machine-learning
# Buat dataframe
<code>
import pandas as pd
# List of course topics with chapters
courses_with_chapters = {
"Introduction to Programming with Python": [
"Chapter 1: Getting Started with Python",
"Chapter 2: Variables and Data Types",
"Chapter 3: Control Flow",
"Chapter 4: Functions and Modules",
"Chapter 5: Working with Data Structures"
],
"Advanced Java Programming": [
"Chapter 1: Advanced Object-Oriented Concepts",
"Chapter 2: Java Collections Framework",
"Chapter 3: Concurrency and Multithreading",
"Chapter 4: Network Programming",
"Chapter 5: Java Memory Management"
],
"Web Development for Beginners": [
"Chapter 1: Introduction to HTML",
"Chapter 2: Styling with CSS",
"Chapter 3: JavaScript Basics",
"Chapter 4: Building Your First Web Page",
"Chapter 5: Deploying a Website"
],
"Mastering Data Structures and Algorithms": [
"Chapter 1: Introduction to Data Structures",
"Chapter 2: Sorting Algorithms",
"Chapter 3: Searching Algorithms",
"Chapter 4: Graph Algorithms",
"Chapter 5: Dynamic Programming"
],
"Fundamentals of Machine Learning": [
"Chapter 1: Introduction to Machine Learning",
"Chapter 2: Supervised Learning",
"Chapter 3: Unsupervised Learning",
"Chapter 4: Neural Networks",
"Chapter 5: Model Evaluation and Validation"
],
"Introduction to Artificial Intelligence": [
"Chapter 1: History of AI",
"Chapter 2: Problem Solving and Search",
"Chapter 3: Knowledge Representation",
"Chapter 4: Machine Learning in AI",
"Chapter 5: Applications of AI"
],
"Building Mobile Apps with Flutter": [
"Chapter 1: Introduction to Flutter",
"Chapter 2: Dart Programming Basics",
"Chapter 3: Building User Interfaces",
"Chapter 4: State Management",
"Chapter 5: Deploying Flutter Apps"
],
"Cybersecurity Essentials": [
"Chapter 1: Understanding Cyber Threats",
"Chapter 2: Network Security",
"Chapter 3: Cryptography Basics",
"Chapter 4: Security Policies and Procedures",
"Chapter 5: Incident Response"
],
"Cloud Computing with AWS": [
"Chapter 1: Introduction to Cloud Computing",
"Chapter 2: AWS Services Overview",
"Chapter 3: AWS Management Console",
"Chapter 4: Deploying Applications on AWS",
"Chapter 5: AWS Security Best Practices"
],
"Data Analysis with R": [
"Chapter 1: Introduction to R",
"Chapter 2: Data Wrangling",
"Chapter 3: Data Visualization",
"Chapter 4: Statistical Analysis",
"Chapter 5: Reporting Results"
],
"Big Data Technologies": [
"Chapter 1: Introduction to Big Data",
"Chapter 2: Hadoop Ecosystem",
"Chapter 3: NoSQL Databases",
"Chapter 4: Big Data Processing",
"Chapter 5: Big Data Analytics"
],
"Introduction to Blockchain": [
"Chapter 1: Understanding Blockchain",
"Chapter 2: Blockchain Components",
"Chapter 3: Smart Contracts",
"Chapter 4: Blockchain Applications",
"Chapter 5: Future of Blockchain"
],
"Digital Marketing Strategies": [
"Chapter 1: Digital Marketing Overview",
"Chapter 2: Content Marketing",
"Chapter 3: Social Media Marketing",
"Chapter 4: Email Marketing",
"Chapter 5: Analytics and Measurement"
],
"Social Media Marketing": [
"Chapter 1: Introduction to Social Media Marketing",
"Chapter 2: Creating a Social Media Strategy",
"Chapter 3: Content Creation and Curation",
"Chapter 4: Social Media Advertising",
"Chapter 5: Measuring Social Media Success"
],
"Search Engine Optimization (SEO)": [
"Chapter 1: Understanding SEO",
"Chapter 2: Keyword Research",
"Chapter 3: On-Page SEO",
"Chapter 4: Off-Page SEO",
"Chapter 5: SEO Analytics"
],
"Content Marketing Mastery": [
"Chapter 1: Introduction to Content Marketing",
"Chapter 2: Content Strategy",
"Chapter 3: Content Creation",
"Chapter 4: Content Distribution",
"Chapter 5: Measuring Content Success"
],
"Project Management Fundamentals": [
"Chapter 1: Introduction to Project Management",
"Chapter 2: Project Planning",
"Chapter 3: Project Execution",
"Chapter 4: Project Monitoring and Control",
"Chapter 5: Project Closure"
],
"Agile and Scrum Methodologies": [
"Chapter 1: Introduction to Agile",
"Chapter 2: Scrum Framework",
"Chapter 3: Roles in Scrum",
"Chapter 4: Scrum Events",
"Chapter 5: Scrum Artifacts"
],
"Leadership and Team Management": [
"Chapter 1: Understanding Leadership",
"Chapter 2: Leadership Styles",
"Chapter 3: Team Dynamics",
"Chapter 4: Conflict Resolution",
"Chapter 5: Leading Remote Teams"
],
"Financial Accounting for Beginners": [
"Chapter 1: Introduction to Financial Accounting",
"Chapter 2: The Accounting Cycle",
"Chapter 3: Financial Statements",
"Chapter 4: Recording Financial Transactions",
"Chapter 5: Analyzing Financial Statements"
],
"Business Analytics with Excel": [
"Chapter 1: Introduction to Excel for Business Analytics",
"Chapter 2: Data Analysis Techniques",
"Chapter 3: Excel Formulas and Functions",
"Chapter 4: Data Visualization with Excel",
"Chapter 5: Advanced Excel Analytics"
],
"Entrepreneurship 101": [
"Chapter 1: Introduction to Entrepreneurship",
"Chapter 2: Business Idea Generation",
"Chapter 3: Business Planning",
"Chapter 4: Marketing for Entrepreneurs",
"Chapter 5: Financial Planning for Startups"
],
"Design Thinking for Innovation": [
"Chapter 1: Introduction to Design Thinking",
"Chapter 2: Empathy in Design",
"Chapter 3: Defining the Problem",
"Chapter 4: Ideation Techniques",
"Chapter 5: Prototyping and Testing"
],
"E-commerce Strategies": [
"Chapter 1: Introduction to E-commerce",
"Chapter 2: E-commerce Business Models",
"Chapter 3: Digital Marketing for E-commerce",
"Chapter 4: E-commerce Website Development",
"Chapter 5: Managing Online Transactions"
],
"Introduction to Graphic Design": [
"Chapter 1: Understanding Graphic Design",
"Chapter 2: Design Principles",
"Chapter 3: Typography",
"Chapter 4: Color Theory",
"Chapter 5: Creating Effective Designs"
],
"Advanced Photoshop Techniques": [
"Chapter 1: Advanced Layer Techniques",
"Chapter 2: Photo Manipulation",
"Chapter 3: Retouching and Restoration",
"Chapter 4: Creating Digital Artwork",
"Chapter 5: Advanced Effects and Filters"
],
"User Experience (UX) Design": [
"Chapter 1: Introduction to UX Design",
"Chapter 2: User Research",
"Chapter 3: Wireframing and Prototyping",
"Chapter 4: Usability Testing",
"Chapter 5: Designing for Accessibility"
],
"3D Modeling with Blender": [
"Chapter 1: Introduction to Blender",
"Chapter 2: Basic Modeling Techniques",
"Chapter 3: Texturing and Shading",
"Chapter 4: Lighting and Rendering",
"Chapter 5: Animation Basics"
],
"Digital Photography": [
"Chapter 1: Introduction to Digital Photography",
"Chapter 2: Camera Basics",
"Chapter 3: Composition Techniques",
"Chapter 4: Lighting in Photography",
"Chapter 5: Post-Processing"
],
"Video Editing with Adobe Premiere Pro": [
"Chapter 1: Introduction to Adobe Premiere Pro",
"Chapter 2: Basic Editing Techniques",
"Chapter 3: Advanced Editing Tools",
"Chapter 4: Adding Effects and Transitions",
"Chapter 5: Exporting and Publishing"
],
"Introduction to Music Production": [
"Chapter 1: Music Production Basics",
"Chapter 2: Digital Audio Workstations (DAWs)",
"Chapter 3: Recording Techniques",
"Chapter 4: Mixing and Mastering",
"Chapter 5: Music Distribution"
],
"Sound Engineering Basics": [
"Chapter 1: Introduction to Sound Engineering",
"Chapter 2: Sound Recording Techniques",
"Chapter 3: Audio Mixing",
"Chapter 4: Live Sound Engineering",
"Chapter 5: Sound Design"
],
"Fundamentals of Game Development": [
"Chapter 1: Introduction to Game Development",
"Chapter 2: Game Design Principles",
"Chapter 3: Game Programming Basics",
"Chapter 4: Graphics and Animation",
"Chapter 5: Testing and Debugging"
],
"Unity Game Development": [
"Chapter 1: Introduction to Unity",
"Chapter 2: Unity Interface and Tools",
"Chapter 3: Scripting in Unity",
"Chapter 4: Physics and Animation in Unity",
"Chapter 5: Building and Publishing Games"
],
"Augmented Reality (AR) Development": [
"Chapter 1: Introduction to AR",
"Chapter 2: AR Development Tools",
"Chapter 3: AR Design Principles",
"Chapter 4: Building AR Applications",
"Chapter 5: Deploying AR Solutions"
],
"Virtual Reality (VR) Development": [
"Chapter 1: Introduction to VR",
"Chapter 2: VR Development Tools",
"Chapter 3: VR Design Principles",
"Chapter 4: Building VR Experiences",
"Chapter 5: Deploying VR Solutions"
],
"Introduction to Robotics": [
"Chapter 1: Understanding Robotics",
"Chapter 2: Robotics Components",
"Chapter 3: Building Simple Robots",
"Chapter 4: Programming Robots",
"Chapter 5: Robotics Applications"
],
"Internet of Things (IoT) Fundamentals": [
"Chapter 1: Introduction to IoT",
"Chapter 2: IoT Architecture",
"Chapter 3: IoT Devices and Sensors",
"Chapter 4: Data Communication in IoT",
"Chapter 5: IoT Applications"
],
"Embedded Systems Design": [
"Chapter 1: Introduction to Embedded Systems",
"Chapter 2: Microcontroller Basics",
"Chapter 3: Embedded Software Development",
"Chapter 4: Interfacing with Hardware",
"Chapter 5: Embedded Systems Applications"
],
"Introduction to Nanotechnology": [
"Chapter 1: Fundamentals of Nanotechnology",
"Chapter 2: Nanomaterials",
"Chapter 3: Nanofabrication Techniques",
"Chapter 4: Applications of Nanotechnology",
"Chapter 5: Future Trends in Nanotechnology"
],
"Renewable Energy Technologies": [
"Chapter 1: Introduction to Renewable Energy",
"Chapter 2: Solar Power",
"Chapter 3: Wind Energy",
"Chapter 4: Hydropower",
"Chapter 5: Energy Storage Solutions"
],
"Sustainable Development Goals": [
"Chapter 1: Understanding Sustainable Development",
"Chapter 2: Overview of the SDGs",
"Chapter 3: Strategies for Achieving the SDGs",
"Chapter 4: Monitoring and Reporting SDGs",
"Chapter 5: Case Studies in Sustainable Development"
],
"Environmental Science": [
"Chapter 1: Introduction to Environmental Science",
"Chapter 2: Ecosystems and Biodiversity",
"Chapter 3: Pollution and Waste Management",
"Chapter 4: Climate Change",
"Chapter 5: Sustainable Resource Management"
],
"Climate Change and Policy": [
"Chapter 1: Understanding Climate Change",
"Chapter 2: Climate Change Science",
"Chapter 3: Climate Policy and Governance",
"Chapter 4: Mitigation and Adaptation Strategies",
"Chapter 5: Global Climate Agreements"
],
"Introduction to Psychology": [
"Chapter 1: Foundations of Psychology",
"Chapter 2: Biological Bases of Behavior",
"Chapter 3: Learning and Memory",
"Chapter 4: Personality and Behavior",
"Chapter 5: Psychological Disorders"
],
"Child Development and Psychology": [
"Chapter 1: Introduction to Child Development",
"Chapter 2: Cognitive Development",
"Chapter 3: Social and Emotional Development",
"Chapter 4: Language Development",
"Chapter 5: Developmental Challenges"
],
"Cognitive Behavioral Therapy (CBT)": [
"Chapter 1: Introduction to CBT",
"Chapter 2: Core Principles of CBT",
"Chapter 3: Cognitive Techniques",
"Chapter 4: Behavioral Techniques",
"Chapter 5: Applications of CBT"
],
"Positive Psychology": [
"Chapter 1: Introduction to Positive Psychology",
"Chapter 2: Happiness and Well-being",
"Chapter 3: Strengths and Virtues",
"Chapter 4: Positive Relationships",
"Chapter 5: Resilience and Flourishing"
],
"Introduction to Sociology": [
"Chapter 1: Foundations of Sociology",
"Chapter 2: Social Institutions",
"Chapter 3: Social Stratification",
"Chapter 4: Social Change",
"Chapter 5: Research Methods in Sociology"
],
"Cultural Anthropology": [
"Chapter 1: Introduction to Cultural Anthropology",
"Chapter 2: Culture and Society",
"Chapter 3: Kinship and Social Organization",
"Chapter 4: Religion and Belief Systems",
"Chapter 5: Globalization and Culture"
],
"World History Overview": [
"Chapter 1: Ancient Civilizations",
"Chapter 2: Medieval Period",
"Chapter 3: Renaissance and Reformation",
"Chapter 4: Industrial Revolution",
"Chapter 5: Modern History"
],
"Ancient Civilizations": [
"Chapter 1: Mesopotamia",
"Chapter 2: Ancient Egypt",
"Chapter 3: Indus Valley",
"Chapter 4: Ancient China",
"Chapter 5: Classical Greece and Rome"
],
"Modern Political Theory": [
"Chapter 1: Foundations of Political Theory",
"Chapter 2: Liberalism and Conservatism",
"Chapter 3: Marxism and Socialism",
"Chapter 4: Feminism and Identity Politics",
"Chapter 5: Global Political Thought"
],
"International Relations": [
"Chapter 1: Introduction to International Relations",
"Chapter 2: Theories of International Relations",
"Chapter 3: International Organizations",
"Chapter 4: Global Security",
"Chapter 5: International Political Economy"
],
"Introduction to Philosophy": [
"Chapter 1: Foundations of Philosophy",
"Chapter 2: Metaphysics",
"Chapter 3: Epistemology",
"Chapter 4: Ethics",
"Chapter 5: Political Philosophy"
],
"Ethics and Moral Philosophy": [
"Chapter 1: Introduction to Ethics",
"Chapter 2: Theories of Moral Philosophy",
"Chapter 3: Applied Ethics",
"Chapter 4: Moral Dilemmas",
"Chapter 5: Ethics in Professional Life"
],
"Creative Writing Workshop": [
"Chapter 1: Fundamentals of Creative Writing",
"Chapter 2: Fiction Writing Techniques",
"Chapter 3: Poetry Writing",
"Chapter 4: Writing for Stage and Screen",
"Chapter 5: Revising and Editing"
],
"Journalism and Media Studies": [
"Chapter 1: Introduction to Journalism",
"Chapter 2: News Writing and Reporting",
"Chapter 3: Media Ethics and Law",
"Chapter 4: Broadcast Journalism",
"Chapter 5: Digital Media"
],
"Public Speaking and Presentation Skills": [
"Chapter 1: Fundamentals of Public Speaking",
"Chapter 2: Crafting Your Message",
"Chapter 3: Vocal and Physical Delivery",
"Chapter 4: Handling Q&A Sessions",
"Chapter 5: Overcoming Public Speaking Anxiety"
],
"Effective Communication Strategies": [
"Chapter 1: Introduction to Communication",
"Chapter 2: Verbal and Nonverbal Communication",
"Chapter 3: Interpersonal Communication",
"Chapter 4: Communication in Teams",
"Chapter 5: Conflict Resolution"
],
"Negotiation and Conflict Resolution": [
"Chapter 1: Fundamentals of Negotiation",
"Chapter 2: Negotiation Strategies",
"Chapter 3: Conflict Resolution Techniques",
"Chapter 4: Mediation and Facilitation",
"Chapter 5: Practical Negotiation Scenarios"
],
"Introduction to Law": [
"Chapter 1: Foundations of Law",
"Chapter 2: Civil Law",
"Chapter 3: Criminal Law",
"Chapter 4: Constitutional Law",
"Chapter 5: International Law"
],
"Intellectual Property Law": [
"Chapter 1: Introduction to Intellectual Property",
"Chapter 2: Copyright Law",
"Chapter 3: Patent Law",
"Chapter 4: Trademark Law",
"Chapter 5: IP Enforcement"
],
"Human Rights and International Law": [
"Chapter 1: Introduction to Human Rights",
"Chapter 2: International Human Rights Law",
"Chapter 3: Human Rights Institutions",
"Chapter 4: Human Rights Violations",
"Chapter 5: Advocacy and Enforcement"
],
"Health and Wellness Coaching": [
"Chapter 1: Introduction to Health Coaching",
"Chapter 2: Wellness Assessment",
"Chapter 3: Behavior Change Techniques",
"Chapter 4: Developing Wellness Plans",
"Chapter 5: Coaching Skills and Ethics"
],
"Nutrition and Dietetics": [
"Chapter 1: Introduction to Nutrition",
"Chapter 2: Macronutrients and Micronutrients",
"Chapter 3: Nutrition Through Life Stages",
"Chapter 4: Diet Planning and Assessment",
"Chapter 5: Clinical Nutrition"
],
"Introduction to Yoga": [
"Chapter 1: History of Yoga",
"Chapter 2: Basic Yoga Poses",
"Chapter 3: Pranayama (Breathing Techniques)",
"Chapter 4: Meditation and Mindfulness",
"Chapter 5: Yoga for Health and Well-being"
],
"Mindfulness and Meditation": [
"Chapter 1: Introduction to Mindfulness",
"Chapter 2: Meditation Techniques",
"Chapter 3: Mindfulness in Daily Life",
"Chapter 4: Benefits of Mindfulness",
"Chapter 5: Advanced Meditation Practices"
],
"Personal Fitness Training": [
"Chapter 1: Introduction to Personal Training",
"Chapter 2: Anatomy and Physiology",
"Chapter 3: Exercise Programming",
"Chapter 4: Nutrition for Fitness",
"Chapter 5: Client Assessment and Management"
],
"Sports Management": [
"Chapter 1: Introduction to Sports Management",
"Chapter 2: Sports Marketing",
"Chapter 3: Event Management",
"Chapter 4: Sports Law and Ethics",
"Chapter 5: Facility Management"
],
"Culinary Arts Fundamentals": [
"Chapter 1: Introduction to Culinary Arts",
"Chapter 2: Knife Skills and Techniques",
"Chapter 3: Cooking Methods",
"Chapter 4: Baking and Pastry Arts",
"Chapter 5: Plating and Presentation"
],
"Baking and Pastry Arts": [
"Chapter 1: Introduction to Baking",
"Chapter 2: Bread and Yeast Products",
"Chapter 3: Cakes and Pastries",
"Chapter 4: Cookies and Confections",
"Chapter 5: Advanced Decorating Techniques"
],
"Wine Tasting and Sommelier Training": [
"Chapter 1: Introduction to Wine Tasting",
"Chapter 2: Types of Wine",
"Chapter 3: Wine Regions and Terroir",
"Chapter 4: Food and Wine Pairing",
"Chapter 5: Wine Service and Etiquette"
],
"Travel and Tourism Management": [
"Chapter 1: Introduction to Travel and Tourism",
"Chapter 2: Tourism Marketing",
"Chapter 3: Destination Management",
"Chapter 4: Travel Operations",
"Chapter 5: Sustainable Tourism"
],
"Hospitality Management": [
"Chapter 1: Introduction to Hospitality Industry",
"Chapter 2: Hotel Management",
"Chapter 3: Food and Beverage Management",
"Chapter 4: Guest Services",
"Chapter 5: Hospitality Marketing"
],
"Real Estate Investment": [
"Chapter 1: Introduction to Real Estate",
"Chapter 2: Property Valuation",
"Chapter 3: Real Estate Financing",
"Chapter 4: Property Management",
"Chapter 5: Real Estate Market Analysis"
],
"Introduction to Forex Trading": [
"Chapter 1: Basics of Forex Trading",
"Chapter 2: Forex Market Analysis",
"Chapter 3: Trading Strategies",
"Chapter 4: Risk Management",
"Chapter 5: Forex Trading Platforms"
],
"Stock Market Investing": [
"Chapter 1: Introduction to Stock Markets",
"Chapter 2: Fundamental Analysis",
"Chapter 3: Technical Analysis",
"Chapter 4: Investment Strategies",
"Chapter 5: Portfolio Management"
],
"Cryptocurrency Trading": [
"Chapter 1: Introduction to Cryptocurrencies",
"Chapter 2: Blockchain Technology",
"Chapter 3: Trading Cryptocurrencies",
"Chapter 4: Crypto Wallets and Exchanges",
"Chapter 5: Crypto Regulations and Security"
],
"Personal Finance Management": [
"Chapter 1: Introduction to Personal Finance",
"Chapter 2: Budgeting and Saving",
"Chapter 3: Debt Management",
"Chapter 4: Investing Basics",
"Chapter 5: Retirement Planning"
],
"Retirement Planning": [
"Chapter 1: Introduction to Retirement Planning",
"Chapter 2: Social Security Benefits",
"Chapter 3: Retirement Savings Plans",
"Chapter 4: Investment Strategies for Retirement",
"Chapter 5: Managing Retirement Income"
],
"Introduction to Astronomy": [
"Chapter 1: Basics of Astronomy",
"Chapter 2: Solar System",
"Chapter 3: Stars and Galaxies",
"Chapter 4: Cosmology",
"Chapter 5: Observational Astronomy"
],
"Astrophysics Fundamentals": [
"Chapter 1: Introduction to Astrophysics",
"Chapter 2: Stellar Physics",
"Chapter 3: Galactic Dynamics",
"Chapter 4: Cosmology",
"Chapter 5: Astrophysical Observations"
],
"Quantum Mechanics": [
"Chapter 1: Introduction to Quantum Mechanics",
"Chapter 2: Quantum States and Operators",
"Chapter 3: Quantum Dynamics",
"Chapter 4: Quantum Field Theory",
"Chapter 5: Applications of Quantum Mechanics"
],
"Introduction to Chemistry": [
"Chapter 1: Basics of Chemistry",
"Chapter 2: Atomic Structure",
"Chapter 3: Chemical Bonding",
"Chapter 4: Chemical Reactions",
"Chapter 5: Organic Chemistry"
],
"Organic Chemistry": [
"Chapter 1: Introduction to Organic Chemistry",
"Chapter 2: Structure and Bonding in Organic Molecules",
"Chapter 3: Organic Reactions and Mechanisms",
"Chapter 4: Synthesis of Organic Compounds",
"Chapter 5: Spectroscopy and Analysis"
],
"Molecular Biology": [
"Chapter 1: Introduction to Molecular Biology",
"Chapter 2: DNA and RNA Structure",
"Chapter 3: Protein Synthesis",
"Chapter 4: Gene Regulation",
"Chapter 5: Molecular Genetics"
],
"Genetics and Genomics": [
"Chapter 1: Introduction to Genetics",
"Chapter 2: Mendelian Genetics",
"Chapter 3: Molecular Genetics",
"Chapter 4: Genomics",
"Chapter 5: Genetic Engineering"
],
"Introduction to Biotechnology": [
"Chapter 1: Basics of Biotechnology",
"Chapter 2: Genetic Engineering",
"Chapter 3: Biotechnology Applications",
"Chapter 4: Bioprocessing",
"Chapter 5: Regulatory and Ethical Issues"
],
"Medical Terminology": [
"Chapter 1: Basics of Medical Terminology",
"Chapter 2: Body Systems and Terminology",
"Chapter 3: Diagnostic Terms",
"Chapter 4: Procedural Terms",
"Chapter 5: Pharmacological Terms"
],
"Pharmacology Basics": [
"Chapter 1: Introduction to Pharmacology",
"Chapter 2: Pharmacokinetics",
"Chapter 3: Pharmacodynamics",
"Chapter 4: Drug Classes and Mechanisms",
"Chapter 5: Clinical Pharmacology"
],
"Nursing Fundamentals": [
"Chapter 1: Introduction to Nursing",
"Chapter 2: Nursing Ethics and Law",
"Chapter 3: Patient Care",
"Chapter 4: Clinical Skills",
"Chapter 5: Nursing Specialties"
],
"Health Informatics": [
"Chapter 1: Introduction to Health Informatics",
"Chapter 2: Health Information Systems",
"Chapter 3: Electronic Health Records (EHR)",
"Chapter 4: Data Standards and Interoperability",
"Chapter 5: Health Data Analytics"
],
"Public Health Principles": [
"Chapter 1: Introduction to Public Health",
"Chapter 2: Epidemiology",
"Chapter 3: Health Policy and Management",
"Chapter 4: Environmental Health",
"Chapter 5: Global Health"
],
"Veterinary Science Basics": [
"Chapter 1: Introduction to Veterinary Science",
"Chapter 2: Animal Anatomy and Physiology",
"Chapter 3: Veterinary Pharmacology",
"Chapter 4: Animal Diseases and Treatments",
"Chapter 5: Veterinary Clinical Practices"
],
"Animal Behavior and Welfare": [
"Chapter 1: Introduction to Animal Behavior",
"Chapter 2: Behavioral Ecology",
"Chapter 3: Animal Cognition",
"Chapter 4: Animal Welfare Science",
"Chapter 5: Practical Animal Welfare"
],
"Horticulture and Plant Science": [
"Chapter 1: Basics of Horticulture",
"Chapter 2: Plant Physiology",
"Chapter 3: Soil Science",
"Chapter 4: Plant Nutrition",
"Chapter 5: Sustainable Horticulture"
],
"Soil Science and Management": [
"Chapter 1: Introduction to Soil Science",
"Chapter 2: Soil Composition and Properties",
"Chapter 3: Soil Fertility and Management",
"Chapter 4: Soil Conservation",
"Chapter 5: Soil Testing and Analysis"
],
"Agricultural Business Management": [
"Chapter 1: Introduction to Agribusiness",
"Chapter 2: Farm Management",
"Chapter 3: Agricultural Marketing",
"Chapter 4: Agricultural Finance",
"Chapter 5: Agricultural Policy"
],
"Introduction to Marine Biology": [
"Chapter 1: Basics of Marine Biology",
"Chapter 2: Marine Ecosystems",
"Chapter 3: Marine Organisms",
"Chapter 4: Oceanography",
"Chapter 5: Marine Conservation"
],
"Data Science with Python": [
"Chapter 1: Introduction to Data Science",
"Chapter 2: Data Wrangling with Pandas",
"Chapter 3: Data Visualization with Matplotlib",
"Chapter 4: Statistical Analysis",
"Chapter 5: Machine Learning with Scikit-Learn"
],
"Deep Learning with TensorFlow": [
"Chapter 1: Introduction to Deep Learning",
"Chapter 2: Neural Network Basics",
"Chapter 3: Building Models with TensorFlow",
"Chapter 4: Training and Evaluation",
"Chapter 5: Advanced Topics in Deep Learning"
],
"Natural Language Processing (NLP)": [
"Chapter 1: Introduction to NLP",
"Chapter 2: Text Preprocessing",
"Chapter 3: Sentiment Analysis",
"Chapter 4: Language Models",
"Chapter 5: NLP Applications"
],
"Introduction to Cloud Computing": [
"Chapter 1: Cloud Computing Basics",
"Chapter 2: Cloud Service Models",
"Chapter 3: Cloud Deployment Models",
"Chapter 4: Cloud Security",
"Chapter 5: Cloud Cost Management"
],
"AWS Certified Solutions Architect": [
"Chapter 1: Introduction to AWS",
"Chapter 2: AWS Core Services",
"Chapter 3: Designing AWS Architecture",
"Chapter 4: Security and Compliance",
"Chapter 5: Monitoring and Optimization"
],
"Microsoft Azure Fundamentals": [
"Chapter 1: Introduction to Azure",
"Chapter 2: Azure Core Services",
"Chapter 3: Azure Solutions Architecture",
"Chapter 4: Security and Privacy",
"Chapter 5: Azure Pricing and Support"
],
"Google Cloud Platform (GCP)": [
"Chapter 1: Introduction to GCP",
"Chapter 2: GCP Core Services",
"Chapter 3: Building Applications on GCP",
"Chapter 4: GCP Security",
"Chapter 5: Managing GCP Costs"
],
"DevOps with Docker and Kubernetes": [
"Chapter 1: Introduction to DevOps",
"Chapter 2: Containerization with Docker",
"Chapter 3: Orchestration with Kubernetes",
"Chapter 4: Continuous Integration and Delivery",
"Chapter 5: Monitoring and Logging"
],
"Full Stack Web Development": [
"Chapter 1: Introduction to Full Stack Development",
"Chapter 2: Front-End Development with React",
"Chapter 3: Back-End Development with Node.js",
"Chapter 4: Database Integration",
"Chapter 5: Deployment and Maintenance"
],
"Mobile App Development with React Native": [
"Chapter 1: Introduction to React Native",
"Chapter 2: Setting Up the Environment",
"Chapter 3: Building User Interfaces",
"Chapter 4: State Management",
"Chapter 5: Deploying React Native Apps"
],
"Introduction to Cybersecurity": [
"Chapter 1: Cybersecurity Fundamentals",
"Chapter 2: Network Security",
"Chapter 3: Threats and Vulnerabilities",
"Chapter 4: Security Measures and Controls",
"Chapter 5: Incident Response"
],
"Ethical Hacking and Penetration Testing": [
"Chapter 1: Introduction to Ethical Hacking",
"Chapter 2: Reconnaissance Techniques",
"Chapter 3: Vulnerability Assessment",
"Chapter 4: Exploitation Techniques",
"Chapter 5: Reporting and Mitigation"
],
"Artificial Intelligence for Business": [
"Chapter 1: AI Basics for Business",
"Chapter 2: AI Applications in Business",
"Chapter 3: Implementing AI Solutions",
"Chapter 4: Ethical Considerations in AI",
"Chapter 5: Future Trends in AI"
],
"Blockchain Development with Ethereum": [
"Chapter 1: Introduction to Blockchain",
"Chapter 2: Smart Contracts with Solidity",
"Chapter 3: Building DApps",
"Chapter 4: Security Best Practices",
"Chapter 5: Blockchain Use Cases"
],
"Introduction to Augmented Reality (AR)": [
"Chapter 1: Understanding AR",
"Chapter 2: AR Development Tools",
"Chapter 3: Creating AR Experiences",
"Chapter 4: Interaction Design for AR",
"Chapter 5: Deploying AR Applications"
],
"Virtual Reality (VR) Development with Unity": [
"Chapter 1: Introduction to VR",
"Chapter 2: VR Development in Unity",
"Chapter 3: Creating VR Environments",
"Chapter 4: VR Interaction Techniques",
"Chapter 5: Publishing VR Experiences"
],
"Game Development with Unreal Engine": [
"Chapter 1: Introduction to Unreal Engine",
"Chapter 2: Level Design",
"Chapter 3: Character Development",
"Chapter 4: Game Mechanics",
"Chapter 5: Testing and Optimization"
],
"Introduction to Machine Learning with Python": [
"Chapter 1: Basics of Machine Learning",
"Chapter 2: Supervised Learning Techniques",
"Chapter 3: Unsupervised Learning Techniques",
"Chapter 4: Model Evaluation",
"Chapter 5: Practical Applications"
],
"Data Visualization with Python": [
"Chapter 1: Introduction to Data Visualization",
"Chapter 2: Data Visualization Tools",
"Chapter 3: Creating Charts with Matplotlib",
"Chapter 4: Advanced Visualization with Seaborn",
"Chapter 5: Interactive Plots with Plotly"
],
"Introduction to Quantum Computing": [
"Chapter 1: Basics of Quantum Computing",
"Chapter 2: Quantum Bits and Gates",
"Chapter 3: Quantum Algorithms",
"Chapter 4: Quantum Programming",
"Chapter 5: Quantum Applications"
],
"Advanced SQL for Data Analysis": [
"Chapter 1: SQL Basics Review",
"Chapter 2: Advanced SQL Queries",
"Chapter 3: Working with Subqueries",
"Chapter 4: Window Functions",
"Chapter 5: Performance Tuning"
],
"Introduction to Geospatial Analysis": [
"Chapter 1: Basics of Geospatial Data",
"Chapter 2: GIS Software and Tools",
"Chapter 3: Spatial Data Visualization",
"Chapter 4: Geospatial Analysis Techniques",
"Chapter 5: Applications of Geospatial Analysis"
],
"Introduction to Ethical AI": [
"Chapter 1: Understanding Ethical AI",
"Chapter 2: Bias in AI",
"Chapter 3: Privacy Concerns",
"Chapter 4: Fairness and Accountability",
"Chapter 5: Building Ethical AI Systems"
],
"Digital Transformation Strategies": [
"Chapter 1: Introduction to Digital Transformation",
"Chapter 2: Key Technologies",
"Chapter 3: Business Process Reengineering",
"Chapter 4: Change Management",
"Chapter 5: Case Studies"
],
"Supply Chain Management": [
"Chapter 1: Basics of Supply Chain Management",
"Chapter 2: Demand Planning",
"Chapter 3: Inventory Management",
"Chapter 4: Logistics and Distribution",
"Chapter 5: Supply Chain Optimization"
],
"Introduction to Systems Thinking": [
"Chapter 1: Understanding Systems Thinking",
"Chapter 2: Systems Dynamics",
"Chapter 3: Modeling Complex Systems",
"Chapter 4: Systems Thinking Tools",
"Chapter 5: Applications of Systems Thinking"
],
"Behavioral Economics": [
"Chapter 1: Introduction to Behavioral Economics",
"Chapter 2: Decision-Making Processes",
"Chapter 3: Heuristics and Biases",
"Chapter 4: Behavioral Game Theory",
"Chapter 5: Applications in Policy and Business"
],
"Introduction to Digital Twin Technology": [
"Chapter 1: Basics of Digital Twin Technology",
"Chapter 2: Building Digital Twins",
"Chapter 3: Data Integration",
"Chapter 4: Use Cases and Applications",
"Chapter 5: Future Trends"
],
"Machine Learning for Healthcare": [
"Chapter 1: Introduction to ML in Healthcare",
"Chapter 2: Healthcare Data",
"Chapter 3: Predictive Modeling",
"Chapter 4: Medical Imaging Analysis",
"Chapter 5: Ethical Considerations"
],
"Sustainable Business Practices": [
"Chapter 1: Introduction to Sustainability",
"Chapter 2: Sustainable Business Models",
"Chapter 3: Environmental Impact Assessment",
"Chapter 4: Corporate Social Responsibility",
"Chapter 5: Case Studies"
],
"Introduction to Renewable Energy": [
"Chapter 1: Basics of Renewable Energy",
"Chapter 2: Solar Energy",
"Chapter 3: Wind Energy",
"Chapter 4: Hydroelectric Power",
"Chapter 5: Emerging Technologies"
],
"Introduction to Quantum Physics": [
"Chapter 1: Basics of Quantum Physics",
"Chapter 2: Quantum Mechanics",
"Chapter 3: Quantum Field Theory",
"Chapter 4: Quantum Entanglement",
"Chapter 5: Applications of Quantum Physics"
],
"Digital Art and Animation": [
"Chapter 1: Introduction to Digital Art",
"Chapter 2: Digital Drawing Techniques",
"Chapter 3: Animation Basics",
"Chapter 4: Advanced Animation Techniques",
"Chapter 5: Creating a Digital Art Portfolio"
],
"Introduction to Autonomous Vehicles": [
"Chapter 1: Basics of Autonomous Vehicles",
"Chapter 2: Sensors and Perception",
"Chapter 3: Decision-Making Algorithms",
"Chapter 4: Autonomous Vehicle Software",
"Chapter 5: Ethical and Legal Issues"
],
"Introduction to Cyber Law": [
"Chapter 1: Basics of Cyber Law",
"Chapter 2: Intellectual Property",
"Chapter 3: Data Protection and Privacy",
"Chapter 4: Cybercrime",
"Chapter 5: International Cyber Law"
],
"Introduction to Space Exploration": [
"Chapter 1: History of Space Exploration",
"Chapter 2: Spacecraft and Technology",
"Chapter 3: Human Spaceflight",
"Chapter 4: Robotic Missions",
"Chapter 5: Future of Space Exploration"
],
"Introduction to Behavioral Psychology": [
"Chapter 1: Basics of Behavioral Psychology",
"Chapter 2: Learning and Conditioning",
"Chapter 3: Behavioral Therapies",
"Chapter 4: Cognitive Behavioral Techniques",
"Chapter 5: Applications of Behavioral Psychology"
],
"Introduction to Fashion Design": [
"Chapter 1: Basics of Fashion Design",
"Chapter 2: Fashion Sketching",
"Chapter 3: Textile Selection",
"Chapter 4: Garment Construction",
"Chapter 5: Fashion Marketing"
],
"Introduction to Interior Design": [
"Chapter 1: Basics of Interior Design",
"Chapter 2: Design Principles",
"Chapter 3: Space Planning",
"Chapter 4: Material and Color Selection",
"Chapter 5: Furniture and Accessories"
],
"Advanced Deep Learning Techniques": [
"Chapter 1: Deep Neural Networks",
"Chapter 2: Convolutional Neural Networks",
"Chapter 3: Recurrent Neural Networks",
"Chapter 4: Generative Adversarial Networks",
"Chapter 5: Advanced Applications"
],
"Python for Data Science": [
"Chapter 1: Data Science Basics",
"Chapter 2: Data Manipulation with Pandas",
"Chapter 3: Data Visualization with Matplotlib and Seaborn",
"Chapter 4: Statistical Analysis",
"Chapter 5: Machine Learning with Scikit-Learn"
],
"Blockchain and Cryptocurrency": [
"Chapter 1: Blockchain Basics",
"Chapter 2: Cryptocurrency Fundamentals",
"Chapter 3: Mining and Consensus",
"Chapter 4: Smart Contracts",
"Chapter 5: Future Trends"
],
"Renewable Energy Systems": [
"Chapter 1: Solar Energy",
"Chapter 2: Wind Power",
"Chapter 3: Hydroelectric Energy",
"Chapter 4: Biomass Energy",
"Chapter 5: Energy Storage Solutions"
],
"Cloud Native Development": [
"Chapter 1: Introduction to Cloud Native",
"Chapter 2: Microservices Architecture",
"Chapter 3: Containerization with Docker",
"Chapter 4: Orchestration with Kubernetes",
"Chapter 5: Continuous Integration and Delivery"
],
"Financial Modeling in Excel": [
"Chapter 1: Basic Financial Statements",
"Chapter 2: Forecasting and Projections",
"Chapter 3: Valuation Models",
"Chapter 4: Risk Analysis",
"Chapter 5: Dashboard Creation"
],
"Advanced Cybersecurity Strategies": [
"Chapter 1: Threat Intelligence",
"Chapter 2: Incident Response",
"Chapter 3: Penetration Testing",
"Chapter 4: Cyber Defense Techniques",
"Chapter 5: Security Governance"
],
"AI in Healthcare": [
"Chapter 1: AI Fundamentals in Healthcare",
"Chapter 2: Medical Image Analysis",
"Chapter 3: Predictive Analytics",
"Chapter 4: Natural Language Processing in Healthcare",
"Chapter 5: Ethical Issues in AI"
],
"Game Design and Development": [
"Chapter 1: Game Design Principles",
"Chapter 2: Storytelling and Narrative",
"Chapter 3: Character Development",
"Chapter 4: Level Design",
"Chapter 5: Testing and Deployment"
],
"Advanced JavaScript Frameworks": [
"Chapter 1: Introduction to JavaScript Frameworks",
"Chapter 2: React.js",
"Chapter 3: Vue.js",
"Chapter 4: Angular",
"Chapter 5: Performance Optimization"
],
"Social Media Analytics": [
"Chapter 1: Social Media Metrics",
"Chapter 2: Data Collection Techniques",
"Chapter 3: Analyzing Engagement",
"Chapter 4: Sentiment Analysis",
"Chapter 5: Reporting and Visualization"
],
"Creative Writing Masterclass": [
"Chapter 1: Writing Techniques",
"Chapter 2: Developing Characters",
"Chapter 3: Plot and Structure",
"Chapter 4: Editing and Revision",
"Chapter 5: Publishing Strategies"
],
"Ethical Hacking Fundamentals": [
"Chapter 1: Introduction to Ethical Hacking",
"Chapter 2: Reconnaissance Techniques",
"Chapter 3: Vulnerability Assessment",
"Chapter 4: Exploitation Methods",
"Chapter 5: Reporting and Mitigation"
],
"Project Management for Professionals": [
"Chapter 1: Project Planning",
"Chapter 2: Resource Management",
"Chapter 3: Risk Management",
"Chapter 4: Agile Methodologies",
"Chapter 5: Project Closure"
],
"Photography Techniques": [
"Chapter 1: Camera Settings",
"Chapter 2: Composition and Lighting",
"Chapter 3: Portrait Photography",
"Chapter 4: Landscape Photography",
"Chapter 5: Post-Processing"
],
"Digital Marketing Essentials": [
"Chapter 1: SEO Strategies",
"Chapter 2: Content Marketing",
"Chapter 3: Social Media Marketing",
"Chapter 4: Email Campaigns",
"Chapter 5: Analytics and Metrics"
],
"Behavioral Finance": [
"Chapter 1: Introduction to Behavioral Finance",
"Chapter 2: Psychological Biases",
"Chapter 3: Decision-Making Processes",
"Chapter 4: Market Anomalies",
"Chapter 5: Behavioral Portfolio Management"
],
"Interior Design Concepts": [
"Chapter 1: Design Principles",
"Chapter 2: Space Planning",
"Chapter 3: Color Theory",
"Chapter 4: Furniture and Decor",
"Chapter 5: Lighting Design"
],
"Fashion Marketing": [
"Chapter 1: Market Research",
"Chapter 2: Branding Strategies",
"Chapter 3: Digital Marketing in Fashion",
"Chapter 4: Retail Management",
"Chapter 5: Fashion Public Relations"
],
"Music Production and Mixing": [
"Chapter 1: Music Theory for Producers",
"Chapter 2: Digital Audio Workstations",
"Chapter 3: Recording Techniques",
"Chapter 4: Mixing and Mastering",
"Chapter 5: Music Distribution"
],
"Advanced SQL Techniques": [
"Chapter 1: SQL Query Optimization",
"Chapter 2: Advanced Joins",
"Chapter 3: Window Functions",
"Chapter 4: Common Table Expressions (CTEs)",
"Chapter 5: SQL in Data Warehousing"
],
"Python for Automation": [
"Chapter 1: Scripting Basics",
"Chapter 2: Web Scraping with BeautifulSoup",
"Chapter 3: Automating File Operations",
"Chapter 4: Task Scheduling",
"Chapter 5: Automation with Selenium"
],
"Machine Learning with R": [
"Chapter 1: Introduction to R for ML",
"Chapter 2: Data Preprocessing",
"Chapter 3: Supervised Learning",
"Chapter 4: Unsupervised Learning",
"Chapter 5: Model Evaluation"
],
"Ethical Hacking and Cyber Defense": [
"Chapter 1: Fundamentals of Ethical Hacking",
"Chapter 2: Network Scanning Techniques",
"Chapter 3: System Hacking",
"Chapter 4: Web Application Security",
"Chapter 5: Defensive Strategies"
],
"Financial Analysis with Excel": [
"Chapter 1: Financial Functions in Excel",
"Chapter 2: Building Financial Models",
"Chapter 3: Scenario and Sensitivity Analysis",
"Chapter 4: Visualizing Financial Data",
"Chapter 5: Reporting and Dashboards"
],
"Quantum Computing for Developers": [
"Chapter 1: Quantum Computing Basics",
"Chapter 2: Quantum Algorithms",
"Chapter 3: Quantum Programming with Qiskit",
"Chapter 4: Quantum Error Correction",
"Chapter 5: Future of Quantum Computing"
],
"Web Development with Django": [
"Chapter 1: Introduction to Django",
"Chapter 2: Building a Django Project",
"Chapter 3: Working with Databases",
"Chapter 4: Django Templates and Forms",
"Chapter 5: Deploying Django Applications"
],
"Data Visualization with Tableau": [
"Chapter 1: Getting Started with Tableau",
"Chapter 2: Connecting to Data Sources",
"Chapter 3: Creating Visualizations",
"Chapter 4: Advanced Dashboard Techniques",
"Chapter 5: Sharing and Publishing Workbooks"
],
"Natural Language Processing with Python": [
"Chapter 1: Basics of NLP",
"Chapter 2: Text Preprocessing Techniques",
"Chapter 3: Sentiment Analysis",
"Chapter 4: Named Entity Recognition",
"Chapter 5: NLP with Deep Learning"
],
"AI and Machine Learning in Business": [
"Chapter 1: AI Basics for Business",
"Chapter 2: Data-Driven Decision Making",
"Chapter 3: Implementing AI Solutions",
"Chapter 4: AI in Operations Management",
"Chapter 5: Ethical Considerations in AI"
],
"Robotics Programming with ROS": [
"Chapter 1: Introduction to ROS",
"Chapter 2: ROS Architecture",
"Chapter 3: Programming Robots with ROS",
"Chapter 4: Sensor Integration",
"Chapter 5: Building and Simulating Robots"
],
"Embedded Systems Programming": [
"Chapter 1: Basics of Embedded Systems",
"Chapter 2: Programming Microcontrollers",
"Chapter 3: Real-Time Operating Systems",
"Chapter 4: Hardware Interfacing",
"Chapter 5: Embedded Systems Projects"
],
"Virtual Reality Development with Unity": [
"Chapter 1: Introduction to VR Development",
"Chapter 2: Setting Up Unity for VR",
"Chapter 3: Creating VR Environments",
"Chapter 4: VR Interaction Techniques",
"Chapter 5: Publishing VR Applications"
],
"Cybersecurity Risk Management": [
"Chapter 1: Fundamentals of Risk Management",
"Chapter 2: Identifying Threats and Vulnerabilities",
"Chapter 3: Risk Assessment Techniques",
"Chapter 4: Mitigation Strategies",
"Chapter 5: Developing a Risk Management Plan"
],
"Mobile App Development with Swift": [
"Chapter 1: Introduction to Swift",
"Chapter 2: Building UI with SwiftUI",
"Chapter 3: Networking in Swift",
"Chapter 4: Data Persistence",
"Chapter 5: Deploying to the App Store"
],
"GIS and Remote Sensing": [
"Chapter 1: Introduction to GIS",
"Chapter 2: Remote Sensing Basics",
"Chapter 3: Spatial Data Analysis",
"Chapter 4: Satellite Imagery Interpretation",
"Chapter 5: GIS Applications"
],
"Artificial Intelligence in Healthcare": [
"Chapter 1: AI Applications in Healthcare",
"Chapter 2: Predictive Analytics in Medicine",
"Chapter 3: Medical Image Analysis",
"Chapter 4: AI in Personalized Medicine",
"Chapter 5: Ethical and Regulatory Issues"
],
"Introduction to Cybersecurity": [
"Chapter 1: Understanding Cybersecurity",
"Chapter 2: Cyber Threats and Attack Vectors",
"Chapter 3: Network Security Basics",
"Chapter 4: Secure Coding Practices",
"Chapter 5: Incident Response and Recovery"
],
"Big Data Analytics with Hadoop": [
"Chapter 1: Introduction to Big Data",
"Chapter 2: Hadoop Ecosystem Overview",
"Chapter 3: Data Processing with MapReduce",
"Chapter 4: Data Storage with HDFS",
"Chapter 5: Advanced Analytics with Hive and Pig"
],
"Full-Stack Web Development with MERN": [
"Chapter 1: Introduction to MERN Stack",
"Chapter 2: Building REST APIs with Node.js and Express",
"Chapter 3: Front-End Development with React",
"Chapter 4: Database Integration with MongoDB",
"Chapter 5: Deployment and Maintenance"
],
"Sustainable Architecture": [
"Chapter 1: Principles of Sustainable Design",
"Chapter 2: Energy-Efficient Building Systems",
"Chapter 3: Sustainable Materials",
"Chapter 4: Green Building Certifications",
"Chapter 5: Case Studies in Sustainable Architecture"
],
"Digital Marketing Analytics": [
"Chapter 1: Introduction to Digital Analytics",
"Chapter 2: Web Analytics",
"Chapter 3: Social Media Metrics",
"Chapter 4: Campaign Performance Tracking",
"Chapter 5: Data-Driven Marketing Strategies"
],
"Advanced Android Development": [
"Chapter 1: Advanced UI Design",
"Chapter 2: Networking and APIs",
"Chapter 3: Background Processing",
"Chapter 4: Security Best Practices",
"Chapter 5: Performance Optimization"
],
"Cloud Security": [
"Chapter 1: Introduction to Cloud Security",
"Chapter 2: Identity and Access Management",
"Chapter 3: Data Protection",
"Chapter 4: Network Security in the Cloud",
"Chapter 5: Compliance and Legal Issues"
],
"IoT Security": [
"Chapter 1: Basics of IoT Security",
"Chapter 2: Securing IoT Devices",
"Chapter 3: IoT Network Security",
"Chapter 4: Data Privacy in IoT",
"Chapter 5: IoT Security Challenges and Solutions"
],
"Human-Computer Interaction (HCI)": [
"Chapter 1: Introduction to HCI",
"Chapter 2: Usability Principles",
"Chapter 3: User-Centered Design",
"Chapter 4: Interaction Techniques",
"Chapter 5: Evaluation Methods"
],
"Financial Engineering": [
"Chapter 1: Introduction to Financial Engineering",
"Chapter 2: Derivatives and Risk Management",
"Chapter 3: Quantitative Methods",
"Chapter 4: Portfolio Theory",
"Chapter 5: Financial Modeling"
],
"Advanced Data Mining": [
"Chapter 1: Introduction to Data Mining",
"Chapter 2: Data Preprocessing Techniques",
"Chapter 3: Association and Clustering",
"Chapter 4: Classification and Regression",
"Chapter 5: Advanced Data Mining Applications"
],
"3D Printing and Additive Manufacturing": [
"Chapter 1: Basics of 3D Printing",
"Chapter 2: Materials for 3D Printing",
"Chapter 3: Designing for Additive Manufacturing",
"Chapter 4: 3D Printing Technologies",
"Chapter 5: Applications of 3D Printing"
],
"Renewable Energy Systems": [
"Chapter 1: Solar Energy Systems",
"Chapter 2: Wind Energy Systems",
"Chapter 3: Biomass Energy Systems",
"Chapter 4: Hydro Energy Systems",
"Chapter 5: Energy Storage Solutions"
],
"Advanced SQL Programming": [
"Chapter 1: Advanced Query Techniques",
"Chapter 2: Stored Procedures and Functions",
"Chapter 3: Performance Tuning",
"Chapter 4: Transaction Management",
"Chapter 5: Advanced Data Analysis"
],
"Legal Aspects of Business": [
"Chapter 1: Business Structures",
"Chapter 2: Contract Law",
"Chapter 3: Employment Law",
"Chapter 4: Intellectual Property",
"Chapter 5: International Business Law"
],
"Ethics in Artificial Intelligence": [
"Chapter 1: Understanding AI Ethics",
"Chapter 2: Bias in AI Systems",
"Chapter 3: Privacy and Security",
"Chapter 4: Ethical Decision Making",
"Chapter 5: Regulatory Frameworks"
],
"Bioinformatics": [
"Chapter 1: Introduction to Bioinformatics",
"Chapter 2: Sequence Analysis",
"Chapter 3: Structural Bioinformatics",
"Chapter 4: Functional Genomics",
"Chapter 5: Bioinformatics Tools and Applications"
],
"Urban Planning and Development": [
"Chapter 1: Principles of Urban Planning",
"Chapter 2: Land Use Planning",
"Chapter 3: Transportation Planning",
"Chapter 4: Sustainable Urban Development",
"Chapter 5: Community Development"
],
"Renewable Energy Management": [
"Chapter 1: Renewable Energy Policies",
"Chapter 2: Energy Economics",
"Chapter 3: Project Management",
"Chapter 4: Financing Renewable Projects",
"Chapter 5: Case Studies in Renewable Energy"
],
"Environmental Impact Assessment": [
"Chapter 1: Introduction to Environmental Impact Assessment",
"Chapter 2: EIA Process and Methods",
"Chapter 3: Case Studies",
"Chapter 4: Legal and Policy Framework",
"Chapter 5: Public Participation in EIA"
],
"Climate Change Mitigation": [
"Chapter 1: Science of Climate Change",
"Chapter 2: Mitigation Strategies",
"Chapter 3: Renewable Energy Solutions",
"Chapter 4: Energy Efficiency",
"Chapter 5: Policy and Economic Tools"
],
"Advanced Wireless Communications": [
"Chapter 1: Fundamentals of Wireless Communication",
"Chapter 2: Wireless Network Architectures",
"Chapter 3: Signal Processing Techniques",
"Chapter 4: Advanced Modulation Techniques",
"Chapter 5: Future Trends in Wireless Communication"
],
"Advanced Microeconomics": [
"Chapter 1: Consumer Theory",
"Chapter 2: Theory of the Firm",
"Chapter 3: Market Structure and Competition",
"Chapter 4: Game Theory",
"Chapter 5: Welfare Economics"
],
"Renewable Energy Technology": [
"Chapter 1: Solar Power Systems",
"Chapter 2: Wind Energy Systems",
"Chapter 3: Biomass and Bioenergy",
"Chapter 4: Geothermal Energy",
"Chapter 5: Future Trends in Renewable Energy"
],
"Advanced Financial Accounting": [
"Chapter 1: Financial Reporting Standards",
"Chapter 2: Consolidated Financial Statements",
"Chapter 3: Revenue Recognition",
"Chapter 4: Leases and Pensions",
"Chapter 5: Financial Statement Analysis"
],
"Cybersecurity Threat Intelligence": [
"Chapter 1: Introduction to Threat Intelligence",
"Chapter 2: Threat Intelligence Lifecycle",
"Chapter 3: Threat Data Collection and Analysis",
"Chapter 4: Cyber Threat Hunting",
"Chapter 5: Threat Intelligence Sharing"
],
"Entrepreneurship and Innovation": [
"Chapter 1: Foundations of Entrepreneurship",
"Chapter 2: Business Model Innovation",
"Chapter 3: Funding and Venture Capital",
"Chapter 4: Scaling and Growth Strategies",
"Chapter 5: Exit Strategies"
],
"Advanced Data Structures": [
"Chapter 1: Trees and Graphs",
"Chapter 2: Hash Tables and Heaps",
"Chapter 3: Advanced Graph Algorithms",
"Chapter 4: Dynamic Programming",
"Chapter 5: Algorithm Optimization"
],
"Genomics and Personalized Medicine": [
"Chapter 1: Introduction to Genomics",
"Chapter 2: Genome Sequencing Technologies",
"Chapter 3: Genetic Variations",
"Chapter 4: Personalized Medicine Approaches",
"Chapter 5: Ethical and Social Issues in Genomics"
],
"Smart Grid Technology": [
"Chapter 1: Introduction to Smart Grids",
"Chapter 2: Smart Grid Architectures",
"Chapter 3: Renewable Integration",
"Chapter 4: Advanced Metering Infrastructure",
"Chapter 5: Smart Grid Cybersecurity"
],
"Product Management Essentials": [
"Chapter 1: Role of a Product Manager",
"Chapter 2: Product Lifecycle Management",
"Chapter 3: Market Research and Analysis",
"Chapter 4: Product Development and Launch",
"Chapter 5: Metrics and KPIs"
],
"Advanced Natural Language Processing": [
"Chapter 1: Deep Learning for NLP",
"Chapter 2: Transformer Models",
"Chapter 3: Text Generation and Summarization",
"Chapter 4: Question Answering Systems",
"Chapter 5: Multilingual NLP"
],
"Advanced Web Development with React": [
"Chapter 1: State Management with Redux",
"Chapter 2: Server-Side Rendering with Next.js",
"Chapter 3: Advanced Hooks and Context API",
"Chapter 4: Performance Optimization",
"Chapter 5: Testing and Deployment"
],
"Data Engineering with Apache Spark": [
"Chapter 1: Introduction to Apache Spark",
"Chapter 2: Spark Core and RDDs",
"Chapter 3: Spark SQL and DataFrames",
"Chapter 4: Spark Streaming",
"Chapter 5: Machine Learning with Spark MLlib"
],
"DevOps Engineering": [
"Chapter 1: Introduction to DevOps",
"Chapter 2: Continuous Integration and Delivery",
"Chapter 3: Infrastructure as Code",
"Chapter 4: Monitoring and Logging",
"Chapter 5: Container Orchestration with Kubernetes"
],
"Energy Management Systems": [
"Chapter 1: Fundamentals of Energy Management",
"Chapter 2: Energy Auditing",
"Chapter 3: Energy Efficiency Technologies",
"Chapter 4: Demand Response Strategies",
"Chapter 5: Energy Management Software"
],
"Health Informatics": [
"Chapter 1: Introduction to Health Informatics",
"Chapter 2: Electronic Health Records",
"Chapter 3: Health Information Exchange",
"Chapter 4: Clinical Decision Support Systems",
"Chapter 5: Health Data Analytics"
],
"Mobile Game Development with Unity": [
"Chapter 1: Introduction to Mobile Game Development",
"Chapter 2: Setting Up Unity for Mobile",
"Chapter 3: Mobile Game Mechanics",
"Chapter 4: Touch and Gesture Controls",
"Chapter 5: Optimizing for Performance"
],
"Sustainable Supply Chain Management": [
"Chapter 1: Introduction to Sustainable Supply Chains",
"Chapter 2: Green Logistics",
"Chapter 3: Sustainable Sourcing",
"Chapter 4: Circular Economy",
"Chapter 5: Case Studies in Sustainability"
],
"Urban Sustainability": [
"Chapter 1: Sustainable Urban Development",
"Chapter 2: Green Building Practices",
"Chapter 3: Urban Agriculture",
"Chapter 4: Sustainable Transportation",
"Chapter 5: Resilient Cities"
],
"Renewable Energy Finance": [
"Chapter 1: Introduction to Renewable Energy Finance",
"Chapter 2: Project Financing",
"Chapter 3: Financial Modeling for Renewable Projects",
"Chapter 4: Risk Management",
"Chapter 5: Policy and Incentives"
],
"Marine Conservation": [
"Chapter 1: Marine Ecosystems",
"Chapter 2: Threats to Marine Biodiversity",
"Chapter 3: Conservation Strategies",
"Chapter 4: Marine Protected Areas",
"Chapter 5: Sustainable Fisheries"
],
"Advanced Digital Marketing": [
"Chapter 1: Advanced SEO Techniques",
"Chapter 2: Conversion Rate Optimization",
"Chapter 3: Programmatic Advertising",
"Chapter 4: Influencer Marketing",
"Chapter 5: Data-Driven Marketing"
],
"Agricultural Robotics": [
"Chapter 1: Introduction to Agricultural Robotics",
"Chapter 2: Autonomous Farming Vehicles",
"Chapter 3: Precision Agriculture",
"Chapter 4: Robotic Harvesting",
"Chapter 5: Future of Agricultural Robotics"
],
"Astrobiology": [
"Chapter 1: The Search for Life Beyond Earth",
"Chapter 2: Habitability Criteria",
"Chapter 3: Extremophiles on Earth",
"Chapter 4: Missions to Mars and Beyond",
"Chapter 5: Astrobiology Research"
],
"Behavioral Neuroscience": [
"Chapter 1: Introduction to Behavioral Neuroscience",
"Chapter 2: Neural Mechanisms of Behavior",
"Chapter 3: Brain Development",
"Chapter 4: Neuroplasticity",
"Chapter 5: Behavioral Disorders"
],
"Biomechanics": [
"Chapter 1: Fundamentals of Biomechanics",
"Chapter 2: Musculoskeletal Mechanics",
"Chapter 3: Motion Analysis",
"Chapter 4: Biomechanical Modeling",
"Chapter 5: Applications in Sports and Medicine"
],
"Bioethics": [
"Chapter 1: Introduction to Bioethics",
"Chapter 2: Ethical Issues in Genetics",
"Chapter 3: Medical Ethics",
"Chapter 4: Environmental Bioethics",
"Chapter 5: Emerging Bioethical Issues"
],
"Cognitive Science": [
"Chapter 1: Introduction to Cognitive Science",
"Chapter 2: Perception and Attention",
"Chapter 3: Memory and Learning",
"Chapter 4: Language and Cognition",
"Chapter 5: Cognitive Neuroscience"
],
"Cultural Anthropology": [
"Chapter 1: Basics of Cultural Anthropology",
"Chapter 2: Kinship and Social Organization",
"Chapter 3: Religion and Ritual",
"Chapter 4: Economic Systems",
"Chapter 5: Political Systems"
],
"Educational Psychology": [
"Chapter 1: Introduction to Educational Psychology",
"Chapter 2: Cognitive Development in Education",
"Chapter 3: Learning Theories",
"Chapter 4: Motivation in Education",
"Chapter 5: Classroom Management"
],
"Environmental Chemistry": [
"Chapter 1: Introduction to Environmental Chemistry",
"Chapter 2: Chemistry of the Atmosphere",
"Chapter 3: Soil and Water Chemistry",
"Chapter 4: Pollution and Toxicology",
"Chapter 5: Environmental Analytical Techniques"
],
"Epidemiology": [
"Chapter 1: Introduction to Epidemiology",
"Chapter 2: Study Designs in Epidemiology",
"Chapter 3: Measures of Disease Frequency",
"Chapter 4: Causal Inference in Epidemiology",
"Chapter 5: Applications of Epidemiology"
],
"Forest Management": [
"Chapter 1: Introduction to Forest Management",
"Chapter 2: Silvicultural Systems",
"Chapter 3: Forest Ecology",
"Chapter 4: Sustainable Forest Management",
"Chapter 5: Forest Policy and Economics"
],
"Genetic Counseling": [
"Chapter 1: Introduction to Genetic Counseling",
"Chapter 2: Genetic Testing",
"Chapter 3: Risk Assessment",
"Chapter 4: Ethical Issues in Genetic Counseling",
"Chapter 5: Case Studies"
],
"Marine Biology": [
"Chapter 1: Basics of Marine Biology",
"Chapter 2: Marine Organisms and Ecosystems",
"Chapter 3: Marine Ecology",
"Chapter 4: Human Impacts on Marine Life",
"Chapter 5: Conservation and Management"
],
"Mathematical Biology": [
"Chapter 1: Introduction to Mathematical Biology",
"Chapter 2: Population Dynamics",
"Chapter 3: Modeling Infectious Diseases",
"Chapter 4: Biological Pattern Formation",
"Chapter 5: Computational Biology Techniques"
],
"Microbial Genetics": [
"Chapter 1: Introduction to Microbial Genetics",
"Chapter 2: Gene Structure and Function",
"Chapter 3: Genetic Transfer in Bacteria",
"Chapter 4: Molecular Techniques",
"Chapter 5: Applications in Biotechnology"
],
"Molecular Medicine": [
"Chapter 1: Introduction to Molecular Medicine",
"Chapter 2: Molecular Basis of Disease",
"Chapter 3: Diagnostic Techniques",
"Chapter 4: Therapeutic Strategies",
"Chapter 5: Personalized Medicine"
],
"Nanotechnology": [
"Chapter 1: Basics of Nanotechnology",
"Chapter 2: Nanomaterials",
"Chapter 3: Nanofabrication Techniques",
"Chapter 4: Applications of Nanotechnology",
"Chapter 5: Ethical and Safety Considerations"
],
"Optics and Photonics": [
"Chapter 1: Fundamentals of Optics",
"Chapter 2: Light-Matter Interaction",
"Chapter 3: Optical Systems and Instruments",
"Chapter 4: Photonic Devices",
"Chapter 5: Applications in Communications and Medicine"
],
"Paleoclimatology": [
"Chapter 1: Introduction to Paleoclimatology",
"Chapter 2: Climate Proxies",
"Chapter 3: Ice Cores and Tree Rings",
"Chapter 4: Climate Models",
"Chapter 5: Lessons from Past Climates"
],
"Petroleum Engineering": [
"Chapter 1: Introduction to Petroleum Engineering",
"Chapter 2: Reservoir Engineering",
"Chapter 3: Drilling Engineering",
"Chapter 4: Production Engineering",
"Chapter 5: Enhanced Oil Recovery"
],
"Plant Biotechnology": [
"Chapter 1: Basics of Plant Biotechnology",
"Chapter 2: Plant Tissue Culture",
"Chapter 3: Genetic Engineering in Plants",
"Chapter 4: Applications in Agriculture",
"Chapter 5: Regulatory and Ethical Issues"
],
"Quantum Optics": [
"Chapter 1: Fundamentals of Quantum Optics",
"Chapter 2: Quantum States of Light",
"Chapter 3: Quantum Entanglement",
"Chapter 4: Quantum Information Processing",
"Chapter 5: Experimental Techniques"
],
"Renewable Energy Storage": [
"Chapter 1: Introduction to Energy Storage",
"Chapter 2: Battery Technologies",
"Chapter 3: Supercapacitors",
"Chapter 4: Hydrogen Storage",
"Chapter 5: Applications and Future Trends"
],
"Space Weather": [
"Chapter 1: Basics of Space Weather",
"Chapter 2: Solar Wind and Magnetic Fields",
"Chapter 3: Effects on Earth's Magnetosphere",
"Chapter 4: Space Weather Prediction",
"Chapter 5: Impact on Technology and Society"
],
"Sustainable Fisheries": [
"Chapter 1: Introduction to Sustainable Fisheries",
"Chapter 2: Fishery Management Practices",
"Chapter 3: Aquaculture",
"Chapter 4: Conservation Strategies",
"Chapter 5: Case Studies in Sustainable Fisheries"
],
"Tropical Medicine": [
"Chapter 1: Introduction to Tropical Medicine",
"Chapter 2: Parasitic Diseases",
"Chapter 3: Vector-Borne Diseases",
"Chapter 4: Public Health Interventions",
"Chapter 5: Case Studies in Tropical Medicine"
],
"Aerodynamics": [
"Chapter 1: Basics of Aerodynamics",
"Chapter 2: Fluid Dynamics Principles",
"Chapter 3: Wing and Airfoil Analysis",
"Chapter 4: Aerodynamic Forces",
"Chapter 5: Applications in Aerospace Engineering"
],
"Advanced Robotics": [
"Chapter 1: Robot Kinematics",
"Chapter 2: Robot Dynamics",
"Chapter 3: Motion Planning",
"Chapter 4: Robot Perception",
"Chapter 5: Advanced Control Techniques"
],
"Astrophysics": [
"Chapter 1: Basics of Astrophysics",
"Chapter 2: Stellar Structure and Evolution",
"Chapter 3: Galactic Dynamics",
"Chapter 4: Cosmology",
"Chapter 5: High-Energy Astrophysics"
],
"Behavioral Economics": [
"Chapter 1: Introduction to Behavioral Economics",
"Chapter 2: Cognitive Biases and Heuristics",
"Chapter 3: Decision-Making Under Uncertainty",
"Chapter 4: Behavioral Game Theory",
"Chapter 5: Policy Applications"
],
"Biostatistics": [
"Chapter 1: Basics of Biostatistics",
"Chapter 2: Probability and Distributions",
"Chapter 3: Hypothesis Testing",
"Chapter 4: Regression Analysis",
"Chapter 5: Survival Analysis"
],
"Cryptography": [
"Chapter 1: Introduction to Cryptography",
"Chapter 2: Symmetric Key Cryptography",
"Chapter 3: Public Key Cryptography",
"Chapter 4: Cryptographic Protocols",
"Chapter 5: Cryptanalysis"
],
"Data Privacy": [
"Chapter 1: Basics of Data Privacy",
"Chapter 2: Data Protection Techniques",
"Chapter 3: Privacy Laws and Regulations",
"Chapter 4: Privacy in the Digital Age",
"Chapter 5: Future Challenges in Data Privacy"
],
"Digital Signal Processing": [
"Chapter 1: Introduction to DSP",
"Chapter 2: Discrete-Time Signals and Systems",
"Chapter 3: Fourier Transform",
"Chapter 4: Digital Filters",
"Chapter 5: Applications of DSP"
],
"Ecological Restoration": [
"Chapter 1: Principles of Ecological Restoration",
"Chapter 2: Restoration Techniques",
"Chapter 3: Monitoring and Assessment",
"Chapter 4: Case Studies in Restoration",
"Chapter 5: Policy and Planning"
],
"Energy Economics": [
"Chapter 1: Introduction to Energy Economics",
"Chapter 2: Energy Markets",
"Chapter 3: Energy Policy and Regulation",
"Chapter 4: Renewable Energy Economics",
"Chapter 5: Global Energy Trends"
],
"Environmental Law": [
"Chapter 1: Introduction to Environmental Law",
"Chapter 2: Environmental Regulations",
"Chapter 3: Legal Frameworks for Pollution Control",
"Chapter 4: Natural Resource Management",
"Chapter 5: International Environmental Law"
],
"Geographic Information Systems (GIS)": [
"Chapter 1: Basics of GIS",
"Chapter 2: Spatial Data Models",
"Chapter 3: GIS Analysis Techniques",
"Chapter 4: GIS Software and Tools",
"Chapter 5: Applications of GIS"
],
"Human Genetics": [
"Chapter 1: Basics of Human Genetics",
"Chapter 2: Mendelian Inheritance",
"Chapter 3: Genetic Disorders",
"Chapter 4: Genomic Technologies",
"Chapter 5: Ethical Issues in Genetics"
],
"Industrial Automation": [
"Chapter 1: Introduction to Industrial Automation",
"Chapter 2: Control Systems",
"Chapter 3: PLC Programming",
"Chapter 4: Robotics in Manufacturing",
"Chapter 5: Automation in Industry 4.0"
],
"Infectious Diseases": [
"Chapter 1: Basics of Infectious Diseases",
"Chapter 2: Bacterial Infections",
"Chapter 3: Viral Infections",
"Chapter 4: Fungal and Parasitic Infections",
"Chapter 5: Public Health Strategies"
],
"Machine Learning for Finance": [
"Chapter 1: Introduction to ML in Finance",
"Chapter 2: Predictive Analytics",
"Chapter 3: Algorithmic Trading",
"Chapter 4: Risk Management",
"Chapter 5: Fraud Detection"
],
"Marine Geology": [
"Chapter 1: Basics of Marine Geology",
"Chapter 2: Ocean Basin Formation",
"Chapter 3: Marine Sediments",
"Chapter 4: Plate Tectonics",
"Chapter 5: Marine Resources"
],
"Neuropsychology": [
"Chapter 1: Introduction to Neuropsychology",
"Chapter 2: Brain-Behavior Relationships",
"Chapter 3: Neuropsychological Assessment",
"Chapter 4: Cognitive Disorders",
"Chapter 5: Rehabilitation Techniques"
],
"Oceanography": [
"Chapter 1: Introduction to Oceanography",
"Chapter 2: Physical Oceanography",
"Chapter 3: Chemical Oceanography",
"Chapter 4: Biological Oceanography",
"Chapter 5: Geological Oceanography"
],
"Photonics": [
"Chapter 1: Introduction to Photonics",
"Chapter 2: Photonic Materials and Devices",
"Chapter 3: Waveguides and Optical Fibers",
"Chapter 4: Photonic Crystals",
"Chapter 5: Applications in Communications and Sensing"
],
"Psychopharmacology": [
"Chapter 1: Basics of Psychopharmacology",
"Chapter 2: Neurotransmitter Systems",
"Chapter 3: Drugs for Mental Disorders",
"Chapter 4: Behavioral Effects of Drugs",
"Chapter 5: Addiction and Dependency"
],
"Sociolinguistics": [
"Chapter 1: Introduction to Sociolinguistics",
"Chapter 2: Language Variation",
"Chapter 3: Language and Identity",
"Chapter 4: Language Attitudes and Policies",
"Chapter 5: Multilingualism"
],
"Space Mission Design": [
"Chapter 1: Fundamentals of Space Missions",
"Chapter 2: Mission Planning",
"Chapter 3: Spacecraft Design",
"Chapter 4: Payload and Instrumentation",
"Chapter 5: Mission Operations"
],
"Sustainable Urban Planning": [
"Chapter 1: Principles of Sustainable Urban Planning",
"Chapter 2: Green Infrastructure",
"Chapter 3: Urban Transportation Planning",
"Chapter 4: Smart Cities",
"Chapter 5: Community Engagement"
],
"Tissue Engineering": [
"Chapter 1: Basics of Tissue Engineering",
"Chapter 2: Biomaterials",
"Chapter 3: Cell Culture Techniques",
"Chapter 4: Scaffold Design",
"Chapter 5: Applications in Regenerative Medicine"
],
"Urban Sociology": [
"Chapter 1: Introduction to Urban Sociology",
"Chapter 2: Theories of Urban Development",
"Chapter 3: Social Stratification in Cities",
"Chapter 4: Urbanization and Globalization",
"Chapter 5: Urban Social Movements"
],
"Advanced Artificial Intelligence": [
"Chapter 1: AI Algorithms and Techniques",
"Chapter 2: Deep Learning Architectures",
"Chapter 3: Reinforcement Learning",
"Chapter 4: AI in Robotics",
"Chapter 5: Ethical AI"
],
"Blockchain for Business": [
"Chapter 1: Blockchain Fundamentals",
"Chapter 2: Blockchain Use Cases",
"Chapter 3: Smart Contracts",
"Chapter 4: Blockchain Integration",
"Chapter 5: Blockchain Security"
],
"Comparative Politics": [
"Chapter 1: Introduction to Comparative Politics",
"Chapter 2: Political Systems and Regimes",
"Chapter 3: Electoral Systems",
"Chapter 4: Comparative Political Economy",
"Chapter 5: Political Culture and Participation"
],
"Crisis Management": [
"Chapter 1: Understanding Crisis Management",
"Chapter 2: Risk Assessment",
"Chapter 3: Crisis Communication",
"Chapter 4: Crisis Response Strategies",
"Chapter 5: Post-Crisis Evaluation"
],
"Ecotoxicology": [
"Chapter 1: Principles of Ecotoxicology",
"Chapter 2: Environmental Pollutants",
"Chapter 3: Toxicological Testing",
"Chapter 4: Risk Assessment and Management",
"Chapter 5: Ecotoxicological Case Studies"
],
"Energy Policy": [
"Chapter 1: Fundamentals of Energy Policy",
"Chapter 2: Policy Instruments",
"Chapter 3: Renewable Energy Policy",
"Chapter 4: Energy Efficiency Policy",
"Chapter 5: International Energy Policy"
],
"Food Security": [
"Chapter 1: Introduction to Food Security",
"Chapter 2: Food Production Systems",
"Chapter 3: Food Distribution and Access",
"Chapter 4: Nutrition and Health",
"Chapter 5: Global Food Security Challenges"
],
"Geopolitics": [
"Chapter 1: Introduction to Geopolitics",
"Chapter 2: Geopolitical Theories",
"Chapter 3: Power and Influence",
"Chapter 4: Geopolitical Conflicts",
"Chapter 5: Future Geopolitical Trends"
],
"Hydrology": [
"Chapter 1: Introduction to Hydrology",
"Chapter 2: The Hydrologic Cycle",
"Chapter 3: Surface Water Hydrology",
"Chapter 4: Groundwater Hydrology",
"Chapter 5: Hydrological Modeling"
],
"International Business": [
"Chapter 1: Global Business Environment",
"Chapter 2: International Trade Theories",
"Chapter 3: Foreign Direct Investment",
"Chapter 4: International Business Strategies",
"Chapter 5: Cross-Cultural Management"
],
"Landscape Architecture": [
"Chapter 1: Introduction to Landscape Architecture",
"Chapter 2: Site Analysis and Planning",
"Chapter 3: Sustainable Landscape Design",
"Chapter 4: Landscape Construction",
"Chapter 5: Urban and Rural Landscapes"
],
"Marine Ecology": [
"Chapter 1: Marine Ecosystems",
"Chapter 2: Marine Species Interactions",
"Chapter 3: Coastal Ecology",
"Chapter 4: Marine Conservation Strategies",
"Chapter 5: Human Impact on Marine Ecology"
],
"Neuroethics": [
"Chapter 1: Introduction to Neuroethics",
"Chapter 2: Ethical Issues in Neuroscience",
"Chapter 3: Cognitive Enhancement",
"Chapter 4: Neuroprivacy",
"Chapter 5: Legal Implications of Neuroscience"
],
"Pediatric Nursing": [
"Chapter 1: Fundamentals of Pediatric Nursing",
"Chapter 2: Child Growth and Development",
"Chapter 3: Pediatric Health Assessment",
"Chapter 4: Common Pediatric Conditions",
"Chapter 5: Family-Centered Care"
],
"Renewable Energy Engineering": [
"Chapter 1: Renewable Energy Principles",
"Chapter 2: Solar Energy Systems",
"Chapter 3: Wind Energy Systems",
"Chapter 4: Biomass and Bioenergy",
"Chapter 5: Energy Storage Solutions"
],
"Robotics and Automation": [
"Chapter 1: Fundamentals of Robotics",
"Chapter 2: Sensors and Actuators",
"Chapter 3: Autonomous Systems",
"Chapter 4: Industrial Automation",
"Chapter 5: Future of Robotics"
],
"Social Network Analysis": [
"Chapter 1: Introduction to Social Network Analysis",
"Chapter 2: Network Theory",
"Chapter 3: Network Data Collection and Analysis",
"Chapter 4: Visualization of Social Networks",
"Chapter 5: Applications of Social Network Analysis"
],
"Sustainable Agriculture": [
"Chapter 1: Principles of Sustainable Agriculture",
"Chapter 2: Agroecology",
"Chapter 3: Organic Farming",
"Chapter 4: Integrated Pest Management",
"Chapter 5: Sustainable Food Systems"
],
"Transport Engineering": [
"Chapter 1: Fundamentals of Transport Engineering",
"Chapter 2: Traffic Flow Theory",
"Chapter 3: Transport Planning and Policy",
"Chapter 4: Public Transport Systems",
"Chapter 5: Intelligent Transportation Systems"
],
"Water Resource Management": [
"Chapter 1: Principles of Water Resource Management",
"Chapter 2: Water Supply Systems",
"Chapter 3: Water Quality Management",
"Chapter 4: Integrated Water Resource Management",
"Chapter 5: Policy and Governance"
],
"Advanced Biochemistry": [
"Chapter 1: Protein Structure and Function",
"Chapter 2: Enzyme Kinetics",
"Chapter 3: Metabolic Pathways",
"Chapter 4: Molecular Genetics",
"Chapter 5: Biochemical Techniques"
],
"Aviation Management": [
"Chapter 1: Introduction to Aviation Management",
"Chapter 2: Airline Operations",
"Chapter 3: Airport Management",
"Chapter 4: Aviation Safety and Security",
"Chapter 5: Aviation Law and Regulations"
],
"Climate Science": [
"Chapter 1: Basics of Climate Science",
"Chapter 2: Climate Systems and Feedbacks",
"Chapter 3: Paleoclimatology",
"Chapter 4: Climate Modeling",
"Chapter 5: Climate Change Impacts and Mitigation"
],
"Comparative Literature": [
"Chapter 1: Introduction to Comparative Literature",
"Chapter 2: Literary Theories and Methods",
"Chapter 3: Comparative Analysis of Texts",
"Chapter 4: Themes and Motifs",
"Chapter 5: Cross-Cultural Literary Studies"
],
"Cultural Heritage Management": [
"Chapter 1: Principles of Cultural Heritage Management",
"Chapter 2: Preservation and Conservation",
"Chapter 3: Heritage Tourism",
"Chapter 4: Cultural Policy and Legislation",
"Chapter 5: Case Studies in Heritage Management"
],
"Environmental Microbiology": [
"Chapter 1: Microbial Ecology",
"Chapter 2: Soil and Water Microbiology",
"Chapter 3: Microbial Biogeochemistry",
"Chapter 4: Environmental Biotechnology",
"Chapter 5: Applications in Environmental Management"
],
"Food Science and Technology": [
"Chapter 1: Food Chemistry",
"Chapter 2: Food Processing Technologies",
"Chapter 3: Food Microbiology",
"Chapter 4: Food Safety and Quality Control",
"Chapter 5: Innovations in Food Science"
],
"Gender Studies": [
"Chapter 1: Introduction to Gender Studies",
"Chapter 2: Feminist Theories",
"Chapter 3: Gender and Society",
"Chapter 4: Gender and Sexuality",
"Chapter 5: Intersectionality"
],
"Geotechnical Engineering": [
"Chapter 1: Introduction to Geotechnical Engineering",
"Chapter 2: Soil Mechanics",
"Chapter 3: Foundation Engineering",
"Chapter 4: Slope Stability",
"Chapter 5: Ground Improvement Techniques"
],
"Healthcare Management": [
"Chapter 1: Introduction to Healthcare Management",
"Chapter 2: Healthcare Systems and Policies",
"Chapter 3: Healthcare Operations Management",
"Chapter 4: Quality Improvement in Healthcare",
"Chapter 5: Healthcare Finance and Economics"
],
"Hospitality and Tourism Management": [
"Chapter 1: Fundamentals of Hospitality Management",
"Chapter 2: Tourism Industry Overview",
"Chapter 3: Hospitality Operations",
"Chapter 4: Marketing in Hospitality and Tourism",
"Chapter 5: Sustainable Tourism Practices"
],
"Human Resource Development": [
"Chapter 1: Introduction to HR Development",
"Chapter 2: Training and Development",
"Chapter 3: Performance Management",
"Chapter 4: Organizational Development",
"Chapter 5: Career Development"
],
"Information Systems Management": [
"Chapter 1: Introduction to Information Systems",
"Chapter 2: Systems Analysis and Design",
"Chapter 3: IT Project Management",
"Chapter 4: Database Management",
"Chapter 5: Information Security Management"
],
"Integrated Marketing Communications": [
"Chapter 1: Fundamentals of IMC",
"Chapter 2: Advertising Strategies",
"Chapter 3: Public Relations",
"Chapter 4: Digital Marketing",
"Chapter 5: Measuring IMC Effectiveness"
],
"Marine Biotechnology": [
"Chapter 1: Introduction to Marine Biotechnology",
"Chapter 2: Marine Microorganisms",
"Chapter 3: Marine Natural Products",
"Chapter 4: Biotechnological Applications",
"Chapter 5: Marine Environmental Biotechnology"
],
"Mathematical Modeling": [
"Chapter 1: Basics of Mathematical Modeling",
"Chapter 2: Modeling with Differential Equations",
"Chapter 3: Stochastic Modeling",
"Chapter 4: Computational Modeling",
"Chapter 5: Applications in Science and Engineering"
],
"Medical Imaging": [
"Chapter 1: Principles of Medical Imaging",
"Chapter 2: X-ray Imaging",
"Chapter 3: MRI and CT Scans",
"Chapter 4: Ultrasound Imaging",
"Chapter 5: Advances in Medical Imaging Technologies"
],
"Music Production": [
"Chapter 1: Introduction to Music Production",
"Chapter 2: Recording Techniques",
"Chapter 3: Mixing and Mastering",
"Chapter 4: Music Production Software",
"Chapter 5: The Business of Music Production"
],
"Nanomedicine": [
"Chapter 1: Basics of Nanomedicine",
"Chapter 2: Nanoparticles in Medicine",
"Chapter 3: Drug Delivery Systems",
"Chapter 4: Nanotechnology in Diagnostics",
"Chapter 5: Regulatory and Ethical Issues"
],
"Pharmaceutical Sciences": [
"Chapter 1: Introduction to Pharmaceutical Sciences",
"Chapter 2: Drug Discovery and Development",
"Chapter 3: Pharmacokinetics and Pharmacodynamics",
"Chapter 4: Formulation and Delivery",
"Chapter 5: Clinical Trials"
],
"Planetary Science": [
"Chapter 1: Introduction to Planetary Science",
"Chapter 2: Planetary Geology",
"Chapter 3: Planetary Atmospheres",
"Chapter 4: Moons and Rings",
"Chapter 5: Exoplanets and Habitability"
],
"Public Administration": [
"Chapter 1: Fundamentals of Public Administration",
"Chapter 2: Public Policy Making",
"Chapter 3: Public Budgeting and Finance",
"Chapter 4: Human Resources in the Public Sector",
"Chapter 5: Ethics and Accountability in Public Administration"
],
"Renewable Energy Project Management": [
"Chapter 1: Introduction to Project Management",
"Chapter 2: Project Planning and Scheduling",
"Chapter 3: Risk Management in Renewable Projects",
"Chapter 4: Financial Management",
"Chapter 5: Project Monitoring and Evaluation"
],
"Supply Chain Analytics": [
"Chapter 1: Introduction to Supply Chain Analytics",
"Chapter 2: Data Collection and Management",
"Chapter 3: Predictive Analytics in Supply Chains",
"Chapter 4: Optimization Techniques",
"Chapter 5: Case Studies in Supply Chain Analytics"
],
"Sustainable Water Management": [
"Chapter 1: Principles of Sustainable Water Management",
"Chapter 2: Water Resources Assessment",
"Chapter 3: Water Conservation Techniques",
"Chapter 4: Integrated Water Management",
"Chapter 5: Policy and Governance"
],
"Theoretical Physics": [
"Chapter 1: Foundations of Theoretical Physics",
"Chapter 2: Quantum Mechanics",
"Chapter 3: Relativity Theory",
"Chapter 4: Particle Physics",
"Chapter 5: String Theory and Beyond"
],
"Urban Development": [
"Chapter 1: Introduction to Urban Development",
"Chapter 2: Urban Planning Theories",
"Chapter 3: Housing and Infrastructure",
"Chapter 4: Economic Development in Urban Areas",
"Chapter 5: Sustainable Urban Growth"
],
"Agroecology": [
"Chapter 1: Principles of Agroecology",
"Chapter 2: Soil Health and Management",
"Chapter 3: Sustainable Crop Production",
"Chapter 4: Agroforestry Systems",
"Chapter 5: Case Studies in Agroecology"
],
"Artificial Neural Networks": [
"Chapter 1: Basics of Neural Networks",
"Chapter 2: Training and Optimization",
"Chapter 3: Convolutional Neural Networks",
"Chapter 4: Recurrent Neural Networks",
"Chapter 5: Neural Network Applications"
],
"Biochemical Engineering": [
"Chapter 1: Fundamentals of Biochemical Engineering",
"Chapter 2: Bioreactor Design",
"Chapter 3: Downstream Processing",
"Chapter 4: Enzyme Technology",
"Chapter 5: Industrial Applications"
],
"Climate Policy": [
"Chapter 1: Introduction to Climate Policy",
"Chapter 2: Policy Instruments and Strategies",
"Chapter 3: International Climate Agreements",
"Chapter 4: National Climate Policies",
"Chapter 5: Evaluating Climate Policy"
],
"Conservation Biology": [
"Chapter 1: Principles of Conservation Biology",
"Chapter 2: Biodiversity and Ecosystems",
"Chapter 3: Conservation Strategies",
"Chapter 4: Conservation Genetics",
"Chapter 5: Global Conservation Issues"
],
"Creative Entrepreneurship": [
"Chapter 1: Introduction to Creative Entrepreneurship",
"Chapter 2: Idea Generation and Innovation",
"Chapter 3: Business Planning and Strategy",
"Chapter 4: Marketing for Creative Industries",
"Chapter 5: Funding and Financial Management"
],
"Cyber Physical Systems": [
"Chapter 1: Introduction to Cyber Physical Systems",
"Chapter 2: Sensors and Actuators",
"Chapter 3: Embedded Systems",
"Chapter 4: Communication Networks",
"Chapter 5: Applications and Case Studies"
],
"Data Governance": [
"Chapter 1: Fundamentals of Data Governance",
"Chapter 2: Data Quality Management",
"Chapter 3: Data Policies and Standards",
"Chapter 4: Privacy and Security",
"Chapter 5: Implementing Data Governance"
],
"Ecological Economics": [
"Chapter 1: Introduction to Ecological Economics",
"Chapter 2: Ecosystem Services and Valuation",
"Chapter 3: Sustainable Development",
"Chapter 4: Environmental Policy and Economics",
"Chapter 5: Ecological Footprint Analysis"
],
"Environmental Health": [
"Chapter 1: Basics of Environmental Health",
"Chapter 2: Environmental Toxicology",
"Chapter 3: Occupational Health",
"Chapter 4: Environmental Epidemiology",
"Chapter 5: Health Risk Assessment"
],
"Ethical Leadership": [
"Chapter 1: Foundations of Ethical Leadership",
"Chapter 2: Ethical Decision Making",
"Chapter 3: Leadership Styles and Ethics",
"Chapter 4: Building Ethical Organizations",
"Chapter 5: Case Studies in Ethical Leadership"
],
"Food Policy": [
"Chapter 1: Introduction to Food Policy",
"Chapter 2: Food Security and Nutrition",
"Chapter 3: Agricultural Policy",
"Chapter 4: Trade and Food Policy",
"Chapter 5: Policy Analysis and Evaluation"
],
"Global Health": [
"Chapter 1: Basics of Global Health",
"Chapter 2: Infectious Diseases",
"Chapter 3: Health Systems and Policy",
"Chapter 4: Global Health Initiatives",
"Chapter 5: Emerging Health Challenges"
],
"Green Chemistry": [
"Chapter 1: Principles of Green Chemistry",
"Chapter 2: Sustainable Synthesis",
"Chapter 3: Green Catalysis",
"Chapter 4: Renewable Resources",
"Chapter 5: Case Studies in Green Chemistry"
],
"Humanitarian Logistics": [
"Chapter 1: Basics of Humanitarian Logistics",
"Chapter 2: Supply Chain Management",
"Chapter 3: Emergency Response Logistics",
"Chapter 4: Technology in Humanitarian Logistics",
"Chapter 5: Case Studies"
],
"Industrial Biotechnology": [
"Chapter 1: Introduction to Industrial Biotechnology",
"Chapter 2: Microbial Biotechnology",
"Chapter 3: Bioprocess Engineering",
"Chapter 4: Bioproducts and Biofuels",
"Chapter 5: Industrial Applications"
],
"International Economics": [
"Chapter 1: Fundamentals of International Economics",
"Chapter 2: Trade Theories and Policies",
"Chapter 3: International Finance",
"Chapter 4: Globalization and Economic Integration",
"Chapter 5: Economic Development"
],
"Marine Engineering": [
"Chapter 1: Basics of Marine Engineering",
"Chapter 2: Marine Propulsion Systems",
"Chapter 3: Marine Electrical Systems",
"Chapter 4: Ship Design and Construction",
"Chapter 5: Marine Safety and Environmental Protection"
],
"Medical Biotechnology": [
"Chapter 1: Introduction to Medical Biotechnology",
"Chapter 2: Genetic Engineering",
"Chapter 3: Therapeutic Proteins",
"Chapter 4: Medical Diagnostics",
"Chapter 5: Regulatory and Ethical Issues"
],
"Meteorology": [
"Chapter 1: Basics of Meteorology",
"Chapter 2: Atmospheric Dynamics",
"Chapter 3: Weather Systems",
"Chapter 4: Climate and Weather Forecasting",
"Chapter 5: Meteorological Instruments and Observations"
],
"Molecular Genetics": [
"Chapter 1: Introduction to Molecular Genetics",
"Chapter 2: Gene Structure and Function",
"Chapter 3: Genetic Mutations",
"Chapter 4: Genetic Engineering Techniques",
"Chapter 5: Applications in Medicine and Agriculture"
],
"Nutritional Science": [
"Chapter 1: Basics of Nutritional Science",
"Chapter 2: Macronutrients and Micronutrients",
"Chapter 3: Nutrition Through Life Stages",
"Chapter 4: Diet and Chronic Diseases",
"Chapter 5: Public Health Nutrition"
],
"Paleontology": [
"Chapter 1: Introduction to Paleontology",
"Chapter 2: Fossil Formation and Types",
"Chapter 3: Evolution of Life",
"Chapter 4: Major Extinction Events",
"Chapter 5: Techniques in Paleontology"
],
"Public Health Nutrition": [
"Chapter 1: Fundamentals of Public Health Nutrition",
"Chapter 2: Nutritional Assessment",
"Chapter 3: Community Nutrition Programs",
"Chapter 4: Nutrition Policy and Advocacy",
"Chapter 5: Global Nutrition Challenges"
],
"Renewable Energy Policy": [
"Chapter 1: Introduction to Renewable Energy Policy",
"Chapter 2: Policy Instruments",
"Chapter 3: Renewable Energy Markets",
"Chapter 4: Economic Impacts",
"Chapter 5: Policy Case Studies"
],
"Space Exploration": [
"Chapter 1: History of Space Exploration",
"Chapter 2: Spacecraft and Missions",
"Chapter 3: Human Spaceflight",
"Chapter 4: Robotic Exploration",
"Chapter 5: Future of Space Exploration"
],
"Sport Psychology": [
"Chapter 1: Introduction to Sport Psychology",
"Chapter 2: Motivation and Goal Setting",
"Chapter 3: Mental Training Techniques",
"Chapter 4: Team Dynamics",
"Chapter 5: Performance Enhancement"
],
"Sustainable Business Practices": [
"Chapter 1: Introduction to Sustainability in Business",
"Chapter 2: Corporate Social Responsibility",
"Chapter 3: Sustainable Supply Chains",
"Chapter 4: Green Marketing",
"Chapter 5: Measuring Sustainability Performance"
],
"Translational Medicine": [
"Chapter 1: Introduction to Translational Medicine",
"Chapter 2: Biomarkers and Diagnostics",
"Chapter 3: Drug Development Process",
"Chapter 4: Clinical Trials",
"Chapter 5: Case Studies in Translational Medicine"
],
"Applied Mathematics": [
"Chapter 1: Introduction to Applied Mathematics",
"Chapter 2: Differential Equations",
"Chapter 3: Numerical Methods",
"Chapter 4: Optimization Techniques",
"Chapter 5: Applications in Engineering and Science"
],
"Bioinformatics Algorithms": [
"Chapter 1: Basics of Bioinformatics",
"Chapter 2: Sequence Alignment",
"Chapter 3: Phylogenetic Trees",
"Chapter 4: Protein Structure Prediction",
"Chapter 5: Genomic Data Analysis"
],
"Carbon Management": [
"Chapter 1: Introduction to Carbon Management",
"Chapter 2: Carbon Footprint Analysis",
"Chapter 3: Carbon Reduction Strategies",
"Chapter 4: Carbon Trading and Offsetting",
"Chapter 5: Policy and Regulation"
],
"Comparative Religion": [
"Chapter 1: Introduction to Comparative Religion",
"Chapter 2: Major World Religions",
"Chapter 3: Religious Texts and Traditions",
"Chapter 4: Interfaith Dialogue",
"Chapter 5: Religion in the Modern World"
],
"Cybersecurity Compliance": [
"Chapter 1: Basics of Cybersecurity Compliance",
"Chapter 2: Regulatory Frameworks",
"Chapter 3: Compliance Strategies",
"Chapter 4: Risk Management",
"Chapter 5: Case Studies in Compliance"
],
"Disaster Management": [
"Chapter 1: Introduction to Disaster Management",
"Chapter 2: Risk Assessment and Mitigation",
"Chapter 3: Disaster Response and Recovery",
"Chapter 4: Emergency Planning",
"Chapter 5: Community Resilience"
],
"Educational Technology": [
"Chapter 1: Introduction to Educational Technology",
"Chapter 2: E-Learning Platforms",
"Chapter 3: Instructional Design",
"Chapter 4: Digital Assessment Tools",
"Chapter 5: Future Trends in EdTech"
],
"Environmental Toxicology": [
"Chapter 1: Basics of Environmental Toxicology",
"Chapter 2: Toxicokinetics and Toxicodynamics",
"Chapter 3: Ecotoxicology",
"Chapter 4: Risk Assessment",
"Chapter 5: Case Studies"
],
"Evolutionary Biology": [
"Chapter 1: Introduction to Evolutionary Biology",
"Chapter 2: Mechanisms of Evolution",
"Chapter 3: Speciation and Phylogeny",
"Chapter 4: Evolutionary Genetics",
"Chapter 5: Human Evolution"
],
"Global Supply Chain Management": [
"Chapter 1: Basics of Global Supply Chain Management",
"Chapter 2: Supply Chain Strategy",
"Chapter 3: Logistics and Distribution",
"Chapter 4: Supply Chain Integration",
"Chapter 5: Risk Management"
],
"Healthcare Analytics": [
"Chapter 1: Introduction to Healthcare Analytics",
"Chapter 2: Data Sources and Methods",
"Chapter 3: Predictive Analytics in Healthcare",
"Chapter 4: Health Data Visualization",
"Chapter 5: Case Studies in Healthcare Analytics"
],
"Human Rights Law": [
"Chapter 1: Introduction to Human Rights Law",
"Chapter 2: International Human Rights Frameworks",
"Chapter 3: Civil and Political Rights",
"Chapter 4: Economic, Social, and Cultural Rights",
"Chapter 5: Enforcement and Case Studies"
],
"Industrial Design": [
"Chapter 1: Basics of Industrial Design",
"Chapter 2: Design Process and Methodology",
"Chapter 3: Ergonomics and Human Factors",
"Chapter 4: Materials and Manufacturing",
"Chapter 5: Case Studies in Industrial Design"
],
"International Development": [
"Chapter 1: Introduction to International Development",
"Chapter 2: Development Theories",
"Chapter 3: Global Institutions and Policies",
"Chapter 4: Sustainable Development Goals",
"Chapter 5: Case Studies in Development"
],
"Library Science": [
"Chapter 1: Basics of Library Science",
"Chapter 2: Cataloging and Classification",
"Chapter 3: Information Retrieval",
"Chapter 4: Digital Libraries",
"Chapter 5: Library Management"
],
"Microfinance": [
"Chapter 1: Introduction to Microfinance",
"Chapter 2: Microcredit and Microinsurance",
"Chapter 3: Microfinance Institutions",
"Chapter 4: Impact Assessment",
"Chapter 5: Challenges and Opportunities"
],
"Ocean Engineering": [
"Chapter 1: Basics of Ocean Engineering",
"Chapter 2: Offshore Structures",
"Chapter 3: Marine Hydrodynamics",
"Chapter 4: Coastal Engineering",
"Chapter 5: Ocean Renewable Energy"
],
"Organizational Behavior": [
"Chapter 1: Introduction to Organizational Behavior",
"Chapter 2: Motivation and Leadership",
"Chapter 3: Group Dynamics",
"Chapter 4: Organizational Culture",
"Chapter 5: Change Management"
],
"Peace and Conflict Studies": [
"Chapter 1: Introduction to Peace and Conflict Studies",
"Chapter 2: Conflict Analysis",
"Chapter 3: Conflict Resolution Strategies",
"Chapter 4: Peacebuilding and Reconciliation",
"Chapter 5: Case Studies in Conflict"
],
"Plant Pathology": [
"Chapter 1: Basics of Plant Pathology",
"Chapter 2: Disease Diagnosis",
"Chapter 3: Pathogen Biology",
"Chapter 4: Disease Management",
"Chapter 5: Case Studies in Plant Pathology"
],
"Public Relations": [
"Chapter 1: Introduction to Public Relations",
"Chapter 2: PR Strategies and Tactics",
"Chapter 3: Media Relations",
"Chapter 4: Crisis Communication",
"Chapter 5: PR Campaign Planning"
],
"Renewable Energy Economics": [
"Chapter 1: Basics of Renewable Energy Economics",
"Chapter 2: Cost-Benefit Analysis",
"Chapter 3: Market Dynamics",
"Chapter 4: Policy and Incentives",
"Chapter 5: Case Studies in Renewable Energy"
],
"Social Work": [
"Chapter 1: Introduction to Social Work",
"Chapter 2: Social Work Theories",
"Chapter 3: Community Practice",
"Chapter 4: Child Welfare",
"Chapter 5: Clinical Social Work"
],
"Structural Engineering": [
"Chapter 1: Basics of Structural Engineering",
"Chapter 2: Structural Analysis",
"Chapter 3: Design of Steel Structures",
"Chapter 4: Design of Concrete Structures",
"Chapter 5: Seismic Design"
],
"Sustainable Architecture": [
"Chapter 1: Principles of Sustainable Design",
"Chapter 2: Energy-Efficient Building Systems",
"Chapter 3: Sustainable Materials",
"Chapter 4: Green Building Certifications",
"Chapter 5: Case Studies in Sustainable Architecture"
],
"Transport Logistics": [
"Chapter 1: Introduction to Transport Logistics",
"Chapter 2: Supply Chain Integration",
"Chapter 3: Logistics Network Design",
"Chapter 4: Inventory Management",
"Chapter 5: Transportation Planning and Management"
],
"Urban Agriculture": [
"Chapter 1: Basics of Urban Agriculture",
"Chapter 2: Vertical Farming",
"Chapter 3: Community Gardens",
"Chapter 4: Urban Livestock",
"Chapter 5: Policy and Planning"
],"Aeronautical Engineering": [
"Chapter 1: Introduction to Aeronautics",
"Chapter 2: Aerodynamic Principles",
"Chapter 3: Aircraft Structures",
"Chapter 4: Propulsion Systems",
"Chapter 5: Flight Mechanics"
],
"Agricultural Economics": [
"Chapter 1: Basics of Agricultural Economics",
"Chapter 2: Agricultural Production Economics",
"Chapter 3: Agricultural Marketing",
"Chapter 4: Agricultural Policy",
"Chapter 5: International Trade in Agriculture"
],
"Biomaterials": [
"Chapter 1: Introduction to Biomaterials",
"Chapter 2: Properties of Biomaterials",
"Chapter 3: Biomaterial Applications",
"Chapter 4: Biocompatibility and Toxicity",
"Chapter 5: Future Trends in Biomaterials"
],
"Construction Management": [
"Chapter 1: Basics of Construction Management",
"Chapter 2: Project Planning and Scheduling",
"Chapter 3: Cost Estimation and Control",
"Chapter 4: Construction Site Management",
"Chapter 5: Safety and Quality Management"
],
"Criminology": [
"Chapter 1: Introduction to Criminology",
"Chapter 2: Theories of Crime",
"Chapter 3: Criminal Justice System",
"Chapter 4: Crime Prevention Strategies",
"Chapter 5: Contemporary Issues in Criminology"
],
"Environmental Engineering": [
"Chapter 1: Introduction to Environmental Engineering",
"Chapter 2: Water and Wastewater Treatment",
"Chapter 3: Air Pollution Control",
"Chapter 4: Solid Waste Management",
"Chapter 5: Environmental Impact Assessment"
],
"Fashion Design": [
"Chapter 1: Fundamentals of Fashion Design",
"Chapter 2: Fashion Illustration",
"Chapter 3: Textile Science",
"Chapter 4: Garment Construction",
"Chapter 5: Fashion Marketing and Merchandising"
],
"Food Safety": [
"Chapter 1: Introduction to Food Safety",
"Chapter 2: Foodborne Pathogens",
"Chapter 3: Food Preservation Methods",
"Chapter 4: HACCP and Food Safety Systems",
"Chapter 5: Food Safety Regulations"
],
"Game Development": [
"Chapter 1: Basics of Game Development",
"Chapter 2: Game Design Principles",
"Chapter 3: Game Programming",
"Chapter 4: Game Art and Animation",
"Chapter 5: Game Testing and Deployment"
],
"Global Marketing": [
"Chapter 1: Introduction to Global Marketing",
"Chapter 2: Market Research and Analysis",
"Chapter 3: Global Marketing Strategies",
"Chapter 4: International Marketing Mix",
"Chapter 5: Digital Marketing in a Global Context"
],
"Healthcare Law": [
"Chapter 1: Introduction to Healthcare Law",
"Chapter 2: Patient Rights and Confidentiality",
"Chapter 3: Healthcare Regulations",
"Chapter 4: Medical Malpractice",
"Chapter 5: Ethical Issues in Healthcare Law"
],
"Industrial Engineering": [
"Chapter 1: Basics of Industrial Engineering",
"Chapter 2: Operations Research",
"Chapter 3: Manufacturing Systems",
"Chapter 4: Quality Control",
"Chapter 5: Industrial Safety and Ergonomics"
],
"Interior Design": [
"Chapter 1: Introduction to Interior Design",
"Chapter 2: Design Principles and Elements",
"Chapter 3: Space Planning",
"Chapter 4: Interior Materials and Finishes",
"Chapter 5: Sustainable Interior Design"
],
"Marine Science": [
"Chapter 1: Basics of Marine Science",
"Chapter 2: Oceanography",
"Chapter 3: Marine Ecology",
"Chapter 4: Marine Resources",
"Chapter 5: Conservation and Management"
],
"Material Science": [
"Chapter 1: Introduction to Material Science",
"Chapter 2: Properties of Materials",
"Chapter 3: Material Characterization",
"Chapter 4: Material Processing Techniques",
"Chapter 5: Applications of Material Science"
],
"Microbiology": [
"Chapter 1: Basics of Microbiology",
"Chapter 2: Microbial Structure and Function",
"Chapter 3: Microbial Genetics",
"Chapter 4: Microbial Pathogenesis",
"Chapter 5: Applied Microbiology"
],
"Nanotechnology Engineering": [
"Chapter 1: Basics of Nanotechnology Engineering",
"Chapter 2: Nanomaterials and Their Properties",
"Chapter 3: Nanofabrication Techniques",
"Chapter 4: Applications of Nanotechnology",
"Chapter 5: Ethical and Safety Issues"
],
"Occupational Health and Safety": [
"Chapter 1: Introduction to Occupational Health and Safety",
"Chapter 2: Workplace Hazards",
"Chapter 3: Safety Management Systems",
"Chapter 4: Occupational Health Programs",
"Chapter 5: Regulatory and Legal Framework"
],
"Performing Arts": [
"Chapter 1: Basics of Performing Arts",
"Chapter 2: Acting Techniques",
"Chapter 3: Dance and Movement",
"Chapter 4: Music and Performance",
"Chapter 5: Stagecraft and Production"
],
"Real Estate Development": [
"Chapter 1: Introduction to Real Estate Development",
"Chapter 2: Market Analysis and Feasibility",
"Chapter 3: Financing and Investment",
"Chapter 4: Property Law and Regulation",
"Chapter 5: Project Management in Real Estate"
],
"Renewable Energy Systems": [
"Chapter 1: Basics of Renewable Energy Systems",
"Chapter 2: Solar Energy Systems",
"Chapter 3: Wind Energy Systems",
"Chapter 4: Biomass Energy Systems",
"Chapter 5: Geothermal Energy Systems"
],
"Social Entrepreneurship": [
"Chapter 1: Introduction to Social Entrepreneurship",
"Chapter 2: Social Business Models",
"Chapter 3: Funding and Resources",
"Chapter 4: Measuring Social Impact",
"Chapter 5: Case Studies in Social Entrepreneurship"
],
"Space Science": [
"Chapter 1: Introduction to Space Science",
"Chapter 2: Planetary Science",
"Chapter 3: Stellar and Galactic Astronomy",
"Chapter 4: Cosmology",
"Chapter 5: Space Missions and Exploration"
],
"Speech Pathology": [
"Chapter 1: Basics of Speech Pathology",
"Chapter 2: Anatomy and Physiology of Speech",
"Chapter 3: Speech Disorders and Diagnosis",
"Chapter 4: Speech Therapy Techniques",
"Chapter 5: Case Studies in Speech Pathology"
],
"Sustainable Energy": [
"Chapter 1: Principles of Sustainable Energy",
"Chapter 2: Energy Efficiency",
"Chapter 3: Renewable Energy Technologies",
"Chapter 4: Sustainable Energy Policy",
"Chapter 5: Case Studies in Sustainable Energy"
],
"Veterinary Science": [
"Chapter 1: Basics of Veterinary Science",
"Chapter 2: Animal Anatomy and Physiology",
"Chapter 3: Animal Diseases and Treatment",
"Chapter 4: Veterinary Surgery",
"Chapter 5: Veterinary Practice Management"
],
"Advanced Geometry": [
"Chapter 1: Euclidean Geometry",
"Chapter 2: Non-Euclidean Geometry",
"Chapter 3: Differential Geometry",
"Chapter 4: Algebraic Geometry",
"Chapter 5: Applications of Geometry"
],
"AI Ethics": [
"Chapter 1: Introduction to AI Ethics",
"Chapter 2: Bias and Fairness",
"Chapter 3: Privacy and Security",
"Chapter 4: Ethical Decision Making",
"Chapter 5: Future of Ethical AI"
],
"Art History": [
"Chapter 1: Introduction to Art History",
"Chapter 2: Ancient Art",
"Chapter 3: Medieval Art",
"Chapter 4: Renaissance Art",
"Chapter 5: Modern and Contemporary Art"
],
"Astronomy": [
"Chapter 1: Basics of Astronomy",
"Chapter 2: Solar System",
"Chapter 3: Stars and Galaxies",
"Chapter 4: Cosmology",
"Chapter 5: Observational Astronomy"
],
"Biomechanics": [
"Chapter 1: Fundamentals of Biomechanics",
"Chapter 2: Musculoskeletal Mechanics",
"Chapter 3: Motion Analysis",
"Chapter 4: Biomechanical Modeling",
"Chapter 5: Applications in Sports and Medicine"
],
"Cloud Architecture": [
"Chapter 1: Fundamentals of Cloud Computing",
"Chapter 2: Cloud Service Models",
"Chapter 3: Cloud Security and Compliance",
"Chapter 4: Designing Cloud Architectures",
"Chapter 5: Case Studies in Cloud Architecture"
],
"Cognitive Neuroscience": [
"Chapter 1: Introduction to Cognitive Neuroscience",
"Chapter 2: Brain Imaging Techniques",
"Chapter 3: Neural Basis of Cognition",
"Chapter 4: Cognitive Development",
"Chapter 5: Neuropsychology"
],
"Creative Writing": [
"Chapter 1: Basics of Creative Writing",
"Chapter 2: Fiction Writing Techniques",
"Chapter 3: Poetry Writing",
"Chapter 4: Writing for Stage and Screen",
"Chapter 5: Revising and Editing"
],
"Digital Marketing Analytics": [
"Chapter 1: Introduction to Digital Marketing Analytics",
"Chapter 2: Web Analytics",
"Chapter 3: Social Media Metrics",
"Chapter 4: Campaign Performance Tracking",
"Chapter 5: Data-Driven Marketing Strategies"
],
"Educational Leadership": [
"Chapter 1: Fundamentals of Educational Leadership",
"Chapter 2: Strategic Planning in Education",
"Chapter 3: Leading Educational Change",
"Chapter 4: Building School Culture",
"Chapter 5: Case Studies in Educational Leadership"
],
"Environmental Policy": [
"Chapter 1: Introduction to Environmental Policy",
"Chapter 2: Policy Instruments",
"Chapter 3: Environmental Regulation",
"Chapter 4: Policy Analysis and Evaluation",
"Chapter 5: Global Environmental Policy"
],
"Ethical Hacking": [
"Chapter 1: Introduction to Ethical Hacking",
"Chapter 2: Network Penetration Testing",
"Chapter 3: Web Application Security",
"Chapter 4: Exploit Development",
"Chapter 5: Reporting and Remediation"
],
"Food Science": [
"Chapter 1: Introduction to Food Science",
"Chapter 2: Food Chemistry",
"Chapter 3: Food Microbiology",
"Chapter 4: Food Processing and Preservation",
"Chapter 5: Food Safety and Quality Control"
],
"Genetic Engineering": [
"Chapter 1: Basics of Genetic Engineering",
"Chapter 2: Gene Cloning and Expression",
"Chapter 3: CRISPR and Genome Editing",
"Chapter 4: Applications in Medicine",
"Chapter 5: Ethical and Regulatory Issues"
],
"Green Building": [
"Chapter 1: Principles of Green Building",
"Chapter 2: Sustainable Building Materials",
"Chapter 3: Energy-Efficient Systems",
"Chapter 4: Indoor Environmental Quality",
"Chapter 5: Green Building Certifications"
],
"Hospitality Management": [
"Chapter 1: Fundamentals of Hospitality Management",
"Chapter 2: Hotel Operations",
"Chapter 3: Food and Beverage Management",
"Chapter 4: Tourism and Travel Services",
"Chapter 5: Hospitality Marketing"
],
"Intelligent Transport Systems": [
"Chapter 1: Introduction to Intelligent Transport Systems",
"Chapter 2: Traffic Management",
"Chapter 3: Public Transportation Systems",
"Chapter 4: Vehicle Communication Technologies",
"Chapter 5: Case Studies in ITS"
],
"Marine Ecology": [
"Chapter 1: Basics of Marine Ecology",
"Chapter 2: Marine Ecosystems",
"Chapter 3: Marine Biodiversity",
"Chapter 4: Human Impacts on Marine Life",
"Chapter 5: Conservation Strategies"
],
"Music Theory": [
"Chapter 1: Introduction to Music Theory",
"Chapter 2: Scales and Modes",
"Chapter 3: Harmony and Chords",
"Chapter 4: Rhythm and Meter",
"Chapter 5: Analysis of Musical Forms"
],
"Renewable Energy Technology": [
"Chapter 1: Principles of Renewable Energy",
"Chapter 2: Solar Power Technologies",
"Chapter 3: Wind Energy Technologies",
"Chapter 4: Biomass and Bioenergy",
"Chapter 5: Emerging Renewable Technologies"
],
"Robotic Process Automation": [
"Chapter 1: Basics of Robotic Process Automation",
"Chapter 2: RPA Tools and Technologies",
"Chapter 3: Designing RPA Workflows",
"Chapter 4: Implementing RPA Solutions",
"Chapter 5: Case Studies in RPA"
],
"Social Impact Assessment": [
"Chapter 1: Introduction to Social Impact Assessment",
"Chapter 2: Methods and Techniques",
"Chapter 3: Community Engagement",
"Chapter 4: Monitoring and Evaluation",
"Chapter 5: Case Studies in SIA"
],
"Space Law": [
"Chapter 1: Introduction to Space Law",
"Chapter 2: International Space Treaties",
"Chapter 3: National Space Legislation",
"Chapter 4: Commercial Space Activities",
"Chapter 5: Future Challenges in Space Law"
],
"Sports Management": [
"Chapter 1: Fundamentals of Sports Management",
"Chapter 2: Event Management",
"Chapter 3: Sports Marketing",
"Chapter 4: Athlete Management",
"Chapter 5: Legal and Ethical Issues in Sports"
],
"Sustainable Finance": [
"Chapter 1: Introduction to Sustainable Finance",
"Chapter 2: ESG Criteria",
"Chapter 3: Green Bonds and Investments",
"Chapter 4: Risk Management",
"Chapter 5: Case Studies in Sustainable Finance"
],
"Systems Biology": [
"Chapter 1: Basics of Systems Biology",
"Chapter 2: Network Biology",
"Chapter 3: Computational Modeling",
"Chapter 4: Systems Medicine",
"Chapter 5: Applications and Future Trends"
],
"Urban Planning": [
"Chapter 1: Introduction to Urban Planning",
"Chapter 2: Land Use Planning",
"Chapter 3: Transportation Planning",
"Chapter 4: Urban Design",
"Chapter 5: Policy and Governance"
],
"Viral Immunology": [
"Chapter 1: Basics of Viral Immunology",
"Chapter 2: Virus-Host Interactions",
"Chapter 3: Immune Response to Viral Infections",
"Chapter 4: Vaccines and Antiviral Therapies",
"Chapter 5: Emerging Viral Diseases"
],
"Advanced Algebra": [
"Chapter 1: Polynomial Functions",
"Chapter 2: Rational Expressions",
"Chapter 3: Exponential and Logarithmic Functions",
"Chapter 4: Sequences and Series",
"Chapter 5: Matrices and Determinants"
],
"Archaeology": [
"Chapter 1: Introduction to Archaeology",
"Chapter 2: Archaeological Methods",
"Chapter 3: Analysis of Material Culture",
"Chapter 4: Archaeological Theory",
"Chapter 5: Case Studies in Archaeology"
],
"Astrophysics": [
"Chapter 1: Basics of Astrophysics",
"Chapter 2: Stellar Structure and Evolution",
"Chapter 3: Galactic Dynamics",
"Chapter 4: Cosmology",
"Chapter 5: High-Energy Astrophysics"
],
"Behavioral Finance": [
"Chapter 1: Introduction to Behavioral Finance",
"Chapter 2: Psychological Biases in Investing",
"Chapter 3: Behavioral Portfolio Theory",
"Chapter 4: Market Anomalies",
"Chapter 5: Behavioral Corporate Finance"
],
"Bioethics": [
"Chapter 1: Introduction to Bioethics",
"Chapter 2: Ethical Issues in Genetics",
"Chapter 3: Medical Ethics",
"Chapter 4: Environmental Bioethics",
"Chapter 5: Emerging Bioethical Issues"
],
"Blockchain Development": [
"Chapter 1: Basics of Blockchain Technology",
"Chapter 2: Blockchain Architecture",
"Chapter 3: Smart Contracts",
"Chapter 4: Blockchain Development Platforms",
"Chapter 5: Decentralized Applications (DApps)"
],
"Chemistry of Natural Products": [
"Chapter 1: Introduction to Natural Products",
"Chapter 2: Biosynthesis of Natural Compounds",
"Chapter 3: Alkaloids and Terpenoids",
"Chapter 4: Phenolic Compounds",
"Chapter 5: Applications in Medicine and Agriculture"
],
"Cultural Studies": [
"Chapter 1: Introduction to Cultural Studies",
"Chapter 2: Theories of Culture",
"Chapter 3: Media and Culture",
"Chapter 4: Identity and Representation",
"Chapter 5: Globalization and Culture"
],
"Data Visualization": [
"Chapter 1: Introduction to Data Visualization",
"Chapter 2: Principles of Effective Visualization",
"Chapter 3: Data Visualization Tools",
"Chapter 4: Interactive Visualizations",
"Chapter 5: Case Studies in Data Visualization"
],
"Design Thinking": [
"Chapter 1: Basics of Design Thinking",
"Chapter 2: Empathy and User Research",
"Chapter 3: Ideation Techniques",
"Chapter 4: Prototyping and Testing",
"Chapter 5: Implementing Design Thinking"
],
"Digital Humanities": [
"Chapter 1: Introduction to Digital Humanities",
"Chapter 2: Text Analysis",
"Chapter 3: Digital Archiving",
"Chapter 4: Digital Mapping",
"Chapter 5: Case Studies in Digital Humanities"
],
"Environmental Sociology": [
"Chapter 1: Introduction to Environmental Sociology",
"Chapter 2: Theories of Environmental Sociology",
"Chapter 3: Human-Environment Interaction",
"Chapter 4: Environmental Movements",
"Chapter 5: Environmental Policy and Governance"
],
"Ethnography": [
"Chapter 1: Basics of Ethnography",
"Chapter 2: Fieldwork Methods",
"Chapter 3: Participant Observation",
"Chapter 4: Ethnographic Writing",
"Chapter 5: Ethical Issues in Ethnography"
],
"Forensic Science": [
"Chapter 1: Introduction to Forensic Science",
"Chapter 2: Crime Scene Investigation",
"Chapter 3: Forensic Biology",
"Chapter 4: Forensic Chemistry",
"Chapter 5: Legal Aspects of Forensic Science"
],
"Genomics": [
"Chapter 1: Basics of Genomics",
"Chapter 2: Genome Sequencing Technologies",
"Chapter 3: Functional Genomics",
"Chapter 4: Comparative Genomics",
"Chapter 5: Applications of Genomics"
],
"Historical Linguistics": [
"Chapter 1: Introduction to Historical Linguistics",
"Chapter 2: Language Change Mechanisms",
"Chapter 3: Comparative Method",
"Chapter 4: Language Families",
"Chapter 5: Reconstruction of Proto-Languages"
],
"International Trade Law": [
"Chapter 1: Basics of International Trade Law",
"Chapter 2: World Trade Organization",
"Chapter 3: Trade Agreements",
"Chapter 4: Trade Disputes and Resolutions",
"Chapter 5: Emerging Issues in Trade Law"
],
"Marine Conservation": [
"Chapter 1: Principles of Marine Conservation",
"Chapter 2: Marine Protected Areas",
"Chapter 3: Fisheries Management",
"Chapter 4: Marine Pollution",
"Chapter 5: Climate Change and the Ocean"
],
"Metallurgy": [
"Chapter 1: Introduction to Metallurgy",
"Chapter 2: Physical Metallurgy",
"Chapter 3: Extractive Metallurgy",
"Chapter 4: Mechanical Metallurgy",
"Chapter 5: Applications of Metallurgy"
],
"Neuropsychology": [
"Chapter 1: Basics of Neuropsychology",
"Chapter 2: Brain-Behavior Relationships",
"Chapter 3: Neuropsychological Assessment",
"Chapter 4: Cognitive Disorders",
"Chapter 5: Rehabilitation Techniques"
],
"Philosophy of Mind": [
"Chapter 1: Introduction to Philosophy of Mind",
"Chapter 2: Dualism and Physicalism",
"Chapter 3: Consciousness and Identity",
"Chapter 4: Mental Causation",
"Chapter 5: Contemporary Debates"
],
"Renewable Energy Economics": [
"Chapter 1: Fundamentals of Renewable Energy Economics",
"Chapter 2: Cost-Benefit Analysis",
"Chapter 3: Renewable Energy Markets",
"Chapter 4: Financial Models",
"Chapter 5: Policy and Incentives"
],
"Social Media Analytics": [
"Chapter 1: Introduction to Social Media Analytics",
"Chapter 2: Data Collection Methods",
"Chapter 3: Analyzing Social Media Data",
"Chapter 4: Sentiment Analysis",
"Chapter 5: Case Studies in Social Media Analytics"
],
"Sociology of Education": [
"Chapter 1: Introduction to Sociology of Education",
"Chapter 2: Theories of Education",
"Chapter 3: Education and Social Inequality",
"Chapter 4: Education Policy",
"Chapter 5: Contemporary Issues in Education"
],
"Sports Nutrition": [
"Chapter 1: Basics of Sports Nutrition",
"Chapter 2: Macronutrient Needs",
"Chapter 3: Micronutrients for Athletes",
"Chapter 4: Hydration and Performance",
"Chapter 5: Nutrition Strategies for Training and Competition"
],
"Strategic Management": [
"Chapter 1: Basics of Strategic Management",
"Chapter 2: Competitive Analysis",
"Chapter 3: Strategic Planning",
"Chapter 4: Strategy Implementation",
"Chapter 5: Case Studies in Strategic Management"
],
"Sustainable Agriculture": [
"Chapter 1: Principles of Sustainable Agriculture",
"Chapter 2: Soil Health",
"Chapter 3: Sustainable Crop Production",
"Chapter 4: Integrated Pest Management",
"Chapter 5: Sustainable Livestock Management"
],
"Theoretical Computer Science": [
"Chapter 1: Introduction to Theoretical Computer Science",
"Chapter 2: Automata Theory",
"Chapter 3: Computational Complexity",
"Chapter 4: Formal Languages",
"Chapter 5: Algorithmic Information Theory"
],
"Water Resource Engineering": [
"Chapter 1: Fundamentals of Water Resources",
"Chapter 2: Hydrology and Hydraulics",
"Chapter 3: Water Supply Systems",
"Chapter 4: Wastewater Treatment",
"Chapter 5: Integrated Water Resource Management"
],
"Anthropology": [
"Chapter 1: Introduction to Anthropology",
"Chapter 2: Cultural Anthropology",
"Chapter 3: Biological Anthropology",
"Chapter 4: Archaeology",
"Chapter 5: Linguistic Anthropology"
],
"Applied Physics": [
"Chapter 1: Basics of Applied Physics",
"Chapter 2: Mechanics and Materials",
"Chapter 3: Electromagnetism",
"Chapter 4: Thermodynamics",
"Chapter 5: Quantum Physics"
],
"Behavioral Science": [
"Chapter 1: Introduction to Behavioral Science",
"Chapter 2: Behavioral Research Methods",
"Chapter 3: Human Development",
"Chapter 4: Social Behavior",
"Chapter 5: Behavioral Interventions"
],
"Biophysics": [
"Chapter 1: Basics of Biophysics",
"Chapter 2: Molecular Biophysics",
"Chapter 3: Cellular Biophysics",
"Chapter 4: Biomechanics",
"Chapter 5: Neurobiophysics"
],
"Climate Adaptation": [
"Chapter 1: Introduction to Climate Adaptation",
"Chapter 2: Risk Assessment",
"Chapter 3: Adaptation Strategies",
"Chapter 4: Policy and Governance",
"Chapter 5: Case Studies in Climate Adaptation"
],
"Communication Studies": [
"Chapter 1: Basics of Communication Studies",
"Chapter 2: Interpersonal Communication",
"Chapter 3: Mass Communication",
"Chapter 4: Digital Communication",
"Chapter 5: Communication Theories"
],
"Developmental Psychology": [
"Chapter 1: Introduction to Developmental Psychology",
"Chapter 2: Cognitive Development",
"Chapter 3: Social and Emotional Development",
"Chapter 4: Language Development",
"Chapter 5: Developmental Challenges"
],
"Earth Sciences": [
"Chapter 1: Introduction to Earth Sciences",
"Chapter 2: Geology",
"Chapter 3: Meteorology",
"Chapter 4: Oceanography",
"Chapter 5: Environmental Science"
],
"Environmental Engineering": [
"Chapter 1: Basics of Environmental Engineering",
"Chapter 2: Water and Wastewater Treatment",
"Chapter 3: Air Pollution Control",
"Chapter 4: Solid Waste Management",
"Chapter 5: Environmental Impact Assessment"
],
"Film Studies": [
"Chapter 1: Introduction to Film Studies",
"Chapter 2: Film Theory and Criticism",
"Chapter 3: Film History",
"Chapter 4: Film Production Techniques",
"Chapter 5: Contemporary Cinema"
],
"Geographic Information Systems": [
"Chapter 1: Basics of GIS",
"Chapter 2: Spatial Data Models",
"Chapter 3: GIS Analysis Techniques",
"Chapter 4: GIS Software and Tools",
"Chapter 5: Applications of GIS"
],
"Global Environmental Governance": [
"Chapter 1: Introduction to Global Environmental Governance",
"Chapter 2: International Environmental Agreements",
"Chapter 3: Global Environmental Institutions",
"Chapter 4: Environmental Policy Analysis",
"Chapter 5: Case Studies in Environmental Governance"
],
"Health Economics": [
"Chapter 1: Basics of Health Economics",
"Chapter 2: Health Care Demand and Supply",
"Chapter 3: Health Insurance Markets",
"Chapter 4: Economic Evaluation in Health Care",
"Chapter 5: Health Policy and Economics"
],
"Industrial Automation": [
"Chapter 1: Basics of Industrial Automation",
"Chapter 2: Control Systems",
"Chapter 3: Programmable Logic Controllers",
"Chapter 4: Robotics in Manufacturing",
"Chapter 5: Industrial Internet of Things (IIoT)"
],
"Intellectual Property Rights": [
"Chapter 1: Introduction to Intellectual Property",
"Chapter 2: Copyright Law",
"Chapter 3: Patent Law",
"Chapter 4: Trademark Law",
"Chapter 5: International IP Law"
],
"Literary Theory": [
"Chapter 1: Basics of Literary Theory",
"Chapter 2: Structuralism and Post-Structuralism",
"Chapter 3: Psychoanalytic Criticism",
"Chapter 4: Feminist Literary Theory",
"Chapter 5: Postcolonial Theory"
],
"Medical Imaging": [
"Chapter 1: Principles of Medical Imaging",
"Chapter 2: X-ray Imaging",
"Chapter 3: MRI and CT Scans",
"Chapter 4: Ultrasound Imaging",
"Chapter 5: Advances in Medical Imaging Technologies"
],
"Metaphysics": [
"Chapter 1: Introduction to Metaphysics",
"Chapter 2: The Nature of Reality",
"Chapter 3: Causation and Free Will",
"Chapter 4: Identity and Time",
"Chapter 5: Metaphysical Theories"
],
"Oceanography": [
"Chapter 1: Introduction to Oceanography",
"Chapter 2: Physical Oceanography",
"Chapter 3: Chemical Oceanography",
"Chapter 4: Biological Oceanography",
"Chapter 5: Geological Oceanography"
],
"Paleoclimatology": [
"Chapter 1: Basics of Paleoclimatology",
"Chapter 2: Climate Proxies",
"Chapter 3: Ice Cores and Tree Rings",
"Chapter 4: Marine Sediments",
"Chapter 5: Paleoclimate Modeling"
],
"Pharmaceutical Chemistry": [
"Chapter 1: Introduction to Pharmaceutical Chemistry",
"Chapter 2: Drug Discovery",
"Chapter 3: Drug Design and Development",
"Chapter 4: Pharmacokinetics and Pharmacodynamics",
"Chapter 5: Regulatory Affairs"
],
"Political Philosophy": [
"Chapter 1: Introduction to Political Philosophy",
"Chapter 2: The Social Contract",
"Chapter 3: Justice and Equality",
"Chapter 4: Liberty and Rights",
"Chapter 5: Contemporary Political Thought"
],
"Public Finance": [
"Chapter 1: Basics of Public Finance",
"Chapter 2: Taxation Principles",
"Chapter 3: Government Expenditure",
"Chapter 4: Public Budgeting",
"Chapter 5: Fiscal Policy"
],
"Quantum Computing": [
"Chapter 1: Introduction to Quantum Computing",
"Chapter 2: Quantum Algorithms",
"Chapter 3: Quantum Cryptography",
"Chapter 4: Quantum Hardware",
"Chapter 5: Applications and Future Directions"
],
"Renewable Energy Engineering": [
"Chapter 1: Principles of Renewable Energy",
"Chapter 2: Solar Power Systems",
"Chapter 3: Wind Energy Systems",
"Chapter 4: Biomass Energy Systems",
"Chapter 5: Hydro and Geothermal Energy Systems"
],
"Sociocultural Anthropology": [
"Chapter 1: Introduction to Sociocultural Anthropology",
"Chapter 2: Kinship and Social Organization",
"Chapter 3: Economic Systems",
"Chapter 4: Political Systems",
"Chapter 5: Religion and Belief Systems"
],
"Sociology of Health": [
"Chapter 1: Introduction to Sociology of Health",
"Chapter 2: Health and Illness",
"Chapter 3: Health Care Systems",
"Chapter 4: Health Disparities",
"Chapter 5: Health Policy"
],
"Sustainable Development": [
"Chapter 1: Principles of Sustainable Development",
"Chapter 2: Environmental Sustainability",
"Chapter 3: Social Sustainability",
"Chapter 4: Economic Sustainability",
"Chapter 5: Case Studies in Sustainable Development"
],
"Systems Engineering": [
"Chapter 1: Basics of Systems Engineering",
"Chapter 2: Systems Design and Integration",
"Chapter 3: Systems Modeling and Simulation",
"Chapter 4: Systems Verification and Validation",
"Chapter 5: Case Studies in Systems Engineering"
],
"Theology": [
"Chapter 1: Introduction to Theology",
"Chapter 2: Historical Theology",
"Chapter 3: Systematic Theology",
"Chapter 4: Comparative Theology",
"Chapter 5: Contemporary Theological Issues"
],
"Virology": [
"Chapter 1: Basics of Virology",
"Chapter 2: Virus Structure and Classification",
"Chapter 3: Viral Replication",
"Chapter 4: Pathogenesis of Viral Infections",
"Chapter 5: Vaccines and Antiviral Therapies"
],
"Agricultural Technology": [
"Chapter 1: Introduction to Agricultural Technology",
"Chapter 2: Precision Agriculture",
"Chapter 3: Agricultural Machinery",
"Chapter 4: Biotechnology in Agriculture",
"Chapter 5: Sustainable Agricultural Practices"
],
"Behavioral Economics": [
"Chapter 1: Introduction to Behavioral Economics",
"Chapter 2: Decision Making and Uncertainty",
"Chapter 3: Behavioral Game Theory",
"Chapter 4: Behavioral Finance",
"Chapter 5: Policy Applications of Behavioral Economics"
],
"Biomedical Engineering": [
"Chapter 1: Basics of Biomedical Engineering",
"Chapter 2: Medical Imaging Technologies",
"Chapter 3: Biomechanics and Biomaterials",
"Chapter 4: Biomedical Signal Processing",
"Chapter 5: Innovations in Biomedical Engineering"
],
"Chemical Engineering": [
"Chapter 1: Fundamentals of Chemical Engineering",
"Chapter 2: Chemical Process Analysis",
"Chapter 3: Chemical Reaction Engineering",
"Chapter 4: Transport Phenomena",
"Chapter 5: Process Design and Simulation"
],
"Comparative Politics": [
"Chapter 1: Introduction to Comparative Politics",
"Chapter 2: Political Systems and Regimes",
"Chapter 3: Electoral Systems",
"Chapter 4: Political Parties and Interest Groups",
"Chapter 5: Policy Making and Governance"
],
"Criminal Justice": [
"Chapter 1: Basics of Criminal Justice",
"Chapter 2: Policing and Law Enforcement",
"Chapter 3: The Court System",
"Chapter 4: Corrections and Rehabilitation",
"Chapter 5: Criminal Justice Policy"
],
"Data Mining": [
"Chapter 1: Introduction to Data Mining",
"Chapter 2: Data Preprocessing",
"Chapter 3: Association Rule Mining",
"Chapter 4: Classification and Prediction",
"Chapter 5: Clustering Techniques"
],
"Earthquake Engineering": [
"Chapter 1: Basics of Earthquake Engineering",
"Chapter 2: Seismic Hazard Analysis",
"Chapter 3: Structural Dynamics",
"Chapter 4: Earthquake-Resistant Design",
"Chapter 5: Case Studies in Earthquake Engineering"
],
"Ethnomusicology": [
"Chapter 1: Introduction to Ethnomusicology",
"Chapter 2: Music and Culture",
"Chapter 3: Fieldwork Methods",
"Chapter 4: Music Analysis Techniques",
"Chapter 5: Contemporary Issues in Ethnomusicology"
],
"Food Biotechnology": [
"Chapter 1: Introduction to Food Biotechnology",
"Chapter 2: Genetic Engineering in Food",
"Chapter 3: Fermentation Technology",
"Chapter 4: Food Safety and Biotechnology",
"Chapter 5: Innovations in Food Biotechnology"
],
"Geospatial Analysis": [
"Chapter 1: Introduction to Geospatial Analysis",
"Chapter 2: Remote Sensing Technologies",
"Chapter 3: Geographic Information Systems",
"Chapter 4: Spatial Data Analysis",
"Chapter 5: Applications in Geospatial Analysis"
],
"Hydrogeology": [
"Chapter 1: Basics of Hydrogeology",
"Chapter 2: Groundwater Flow",
"Chapter 3: Aquifer Properties",
"Chapter 4: Groundwater Modeling",
"Chapter 5: Groundwater Management"
],
"Industrial Robotics": [
"Chapter 1: Introduction to Industrial Robotics",
"Chapter 2: Robot Kinematics",
"Chapter 3: Robot Dynamics and Control",
"Chapter 4: Robotic Sensors and Actuators",
"Chapter 5: Applications of Industrial Robotics"
],
"Landscape Ecology": [
"Chapter 1: Basics of Landscape Ecology",
"Chapter 2: Landscape Patterns and Processes",
"Chapter 3: Landscape Modeling",
"Chapter 4: Landscape Conservation",
"Chapter 5: Applications of Landscape Ecology"
],
"Marine Geology": [
"Chapter 1: Introduction to Marine Geology",
"Chapter 2: Ocean Basin Formation",
"Chapter 3: Marine Sediments",
"Chapter 4: Coastal Geomorphology",
"Chapter 5: Marine Geological Hazards"
],
"Meteorology": [
"Chapter 1: Introduction to Meteorology",
"Chapter 2: Atmospheric Dynamics",
"Chapter 3: Weather Systems",
"Chapter 4: Climate and Weather Forecasting",
"Chapter 5: Meteorological Instruments and Observations"
],
"Nanophotonics": [
"Chapter 1: Basics of Nanophotonics",
"Chapter 2: Nanophotonic Materials",
"Chapter 3: Light-Matter Interactions at Nanoscale",
"Chapter 4: Nanophotonic Devices",
"Chapter 5: Applications in Communication and Sensing"
],
"Paleobotany": [
"Chapter 1: Introduction to Paleobotany",
"Chapter 2: Fossil Plants",
"Chapter 3: Paleoecology",
"Chapter 4: Evolution of Plants",
"Chapter 5: Techniques in Paleobotany"
],
"Pharmaceutical Sciences": [
"Chapter 1: Basics of Pharmaceutical Sciences",
"Chapter 2: Drug Discovery and Development",
"Chapter 3: Pharmacokinetics and Pharmacodynamics",
"Chapter 4: Drug Formulation and Delivery",
"Chapter 5: Regulatory Affairs in Pharmaceuticals"
],
"Quantum Optics": [
"Chapter 1: Introduction to Quantum Optics",
"Chapter 2: Quantum States of Light",
"Chapter 3: Interaction of Light with Matter",
"Chapter 4: Quantum Entanglement",
"Chapter 5: Applications in Quantum Communication and Computing"
],
"Renewable Energy Policy": [
"Chapter 1: Introduction to Renewable Energy Policy",
"Chapter 2: Policy Instruments and Strategies",
"Chapter 3: Renewable Energy Markets",
"Chapter 4: Economic Impacts of Renewable Energy",
"Chapter 5: Policy Case Studies"
],
"Social Psychology": [
"Chapter 1: Basics of Social Psychology",
"Chapter 2: Social Perception and Cognition",
"Chapter 3: Attitudes and Persuasion",
"Chapter 4: Group Dynamics",
"Chapter 5: Social Influence and Behavior"
],
"Space Weather": [
"Chapter 1: Introduction to Space Weather",
"Chapter 2: Solar Activity and Phenomena",
"Chapter 3: Earth's Magnetosphere",
"Chapter 4: Space Weather Prediction",
"Chapter 5: Impacts of Space Weather on Technology"
],
"Structural Biology": [
"Chapter 1: Introduction to Structural Biology",
"Chapter 2: Protein Structure Determination",
"Chapter 3: Nucleic Acid Structures",
"Chapter 4: Molecular Interactions",
"Chapter 5: Applications in Drug Design"
],
"Sustainable Construction": [
"Chapter 1: Basics of Sustainable Construction",
"Chapter 2: Green Building Materials",
"Chapter 3: Energy-Efficient Design",
"Chapter 4: Waste Reduction Techniques",
"Chapter 5: Case Studies in Sustainable Construction"
],
"Transportation Engineering": [
"Chapter 1: Basics of Transportation Engineering",
"Chapter 2: Traffic Flow Theory",
"Chapter 3: Transportation Planning",
"Chapter 4: Public Transportation Systems",
"Chapter 5: Intelligent Transportation Systems"
],
"Urban Sociology": [
"Chapter 1: Introduction to Urban Sociology",
"Chapter 2: Urbanization and Urbanism",
"Chapter 3: Social Stratification in Urban Areas",
"Chapter 4: Urban Policy and Planning",
"Chapter 5: Contemporary Urban Issues"
],
"Veterinary Pharmacology": [
"Chapter 1: Basics of Veterinary Pharmacology",
"Chapter 2: Pharmacokinetics in Animals",
"Chapter 3: Drug Therapies for Animals",
"Chapter 4: Veterinary Toxicology",
"Chapter 5: Regulatory Aspects of Veterinary Drugs"
],
"Water Resource Management": [
"Chapter 1: Principles of Water Resource Management",
"Chapter 2: Water Supply Systems",
"Chapter 3: Water Quality Management",
"Chapter 4: Integrated Water Resource Management",
"Chapter 5: Policy and Governance"
],
"Advanced Macroeconomics": [
"Chapter 1: Macroeconomic Theories",
"Chapter 2: Economic Growth",
"Chapter 3: Inflation and Unemployment",
"Chapter 4: Monetary and Fiscal Policy",
"Chapter 5: International Macroeconomics"
],
"Aerodynamics": [
"Chapter 1: Basics of Aerodynamics",
"Chapter 2: Fluid Dynamics",
"Chapter 3: Wing and Airfoil Theory",
"Chapter 4: Supersonic and Hypersonic Flows",
"Chapter 5: Computational Fluid Dynamics"
],
"Astrobiology": [
"Chapter 1: Introduction to Astrobiology",
"Chapter 2: Origin of Life",
"Chapter 3: Extremophiles",
"Chapter 4: Habitable Zones",
"Chapter 5: Search for Extraterrestrial Life"
],
"Biostatistics": [
"Chapter 1: Basics of Biostatistics",
"Chapter 2: Probability Theory",
"Chapter 3: Statistical Inference",
"Chapter 4: Experimental Design",
"Chapter 5: Statistical Methods in Epidemiology"
],
"Carbon Capture and Storage": [
"Chapter 1: Introduction to Carbon Capture",
"Chapter 2: Capture Technologies",
"Chapter 3: Transport of CO2",
"Chapter 4: Storage Techniques",
"Chapter 5: Policy and Economics"
],
"Cognitive Science": [
"Chapter 1: Basics of Cognitive Science",
"Chapter 2: Perception and Cognition",
"Chapter 3: Language and Communication",
"Chapter 4: Cognitive Neuroscience",
"Chapter 5: Artificial Intelligence in Cognitive Science"
],
"Data Ethics": [
"Chapter 1: Introduction to Data Ethics",
"Chapter 2: Privacy and Security",
"Chapter 3: Ethical Data Collection",
"Chapter 4: Bias and Fairness",
"Chapter 5: Ethical Data Use and Governance"
],
"Digital Art": [
"Chapter 1: Basics of Digital Art",
"Chapter 2: Digital Painting Techniques",
"Chapter 3: 3D Modeling and Animation",
"Chapter 4: Digital Photography",
"Chapter 5: Digital Art in Media and Gaming"
],
"Ecotourism": [
"Chapter 1: Introduction to Ecotourism",
"Chapter 2: Principles of Sustainable Tourism",
"Chapter 3: Ecotourism Planning and Development",
"Chapter 4: Ecotourism Management",
"Chapter 5: Case Studies in Ecotourism"
],
"Energy Storage Technologies": [
"Chapter 1: Introduction to Energy Storage",
"Chapter 2: Battery Technologies",
"Chapter 3: Thermal Energy Storage",
"Chapter 4: Mechanical Storage Systems",
"Chapter 5: Emerging Energy Storage Technologies"
],
"Environmental Law": [
"Chapter 1: Basics of Environmental Law",
"Chapter 2: Environmental Regulations",
"Chapter 3: International Environmental Law",
"Chapter 4: Environmental Impact Assessment",
"Chapter 5: Case Studies in Environmental Law"
],
"Fashion Marketing": [
"Chapter 1: Basics of Fashion Marketing",
"Chapter 2: Market Research and Analysis",
"Chapter 3: Branding and Positioning",
"Chapter 4: Digital Marketing in Fashion",
"Chapter 5: Global Fashion Marketing Strategies"
],
"Geophysics": [
"Chapter 1: Introduction to Geophysics",
"Chapter 2: Seismology",
"Chapter 3: Geomagnetism",
"Chapter 4: Gravity and Geodesy",
"Chapter 5: Geophysical Survey Methods"
],
"Historical Archaeology": [
"Chapter 1: Basics of Historical Archaeology",
"Chapter 2: Material Culture Analysis",
"Chapter 3: Archaeological Field Methods",
"Chapter 4: Post-Excavation Analysis",
"Chapter 5: Case Studies in Historical Archaeology"
],
"Hydrology": [
"Chapter 1: Basics of Hydrology",
"Chapter 2: Hydrological Cycle",
"Chapter 3: Surface Water Hydrology",
"Chapter 4: Groundwater Hydrology",
"Chapter 5: Hydrological Modeling"
],
"International Business": [
"Chapter 1: Introduction to International Business",
"Chapter 2: Global Trade Theories",
"Chapter 3: International Business Environment",
"Chapter 4: Global Marketing Strategies",
"Chapter 5: International Business Management"
],
"Marine Biotechnology": [
"Chapter 1: Basics of Marine Biotechnology",
"Chapter 2: Marine Bioresources",
"Chapter 3: Marine Genomics",
"Chapter 4: Marine Bioproducts",
"Chapter 5: Applications and Innovations"
],
"Metrology": [
"Chapter 1: Basics of Metrology",
"Chapter 2: Measurement Standards",
"Chapter 3: Calibration Techniques",
"Chapter 4: Precision and Accuracy",
"Chapter 5: Industrial Applications of Metrology"
],
"Nanomedicine": [
"Chapter 1: Introduction to Nanomedicine",
"Chapter 2: Nanoparticles in Medicine",
"Chapter 3: Drug Delivery Systems",
"Chapter 4: Nanotechnology in Diagnostics",
"Chapter 5: Regulatory and Ethical Issues"
],
"Petroleum Engineering": [
"Chapter 1: Basics of Petroleum Engineering",
"Chapter 2: Reservoir Engineering",
"Chapter 3: Drilling Engineering",
"Chapter 4: Production Engineering",
"Chapter 5: Enhanced Oil Recovery Techniques"
],
"Project Management": [
"Chapter 1: Introduction to Project Management",
"Chapter 2: Project Planning and Scheduling",
"Chapter 3: Risk Management",
"Chapter 4: Project Execution and Control",
"Chapter 5: Project Closure and Evaluation"
],
"Quantum Mechanics": [
"Chapter 1: Basics of Quantum Mechanics",
"Chapter 2: Quantum States and Operators",
"Chapter 3: Quantum Dynamics",
"Chapter 4: Quantum Field Theory",
"Chapter 5: Applications of Quantum Mechanics"
],
"Sustainable Energy Systems": [
"Chapter 1: Basics of Sustainable Energy",
"Chapter 2: Renewable Energy Technologies",
"Chapter 3: Energy Efficiency",
"Chapter 4: Sustainable Energy Policy",
"Chapter 5: Case Studies in Sustainable Energy"
],
"Theoretical Physics": [
"Chapter 1: Introduction to Theoretical Physics",
"Chapter 2: Classical Mechanics",
"Chapter 3: Electrodynamics",
"Chapter 4: Quantum Mechanics",
"Chapter 5: Statistical Mechanics"
],
"Toxicology": [
"Chapter 1: Basics of Toxicology",
"Chapter 2: Toxicokinetics and Toxicodynamics",
"Chapter 3: Environmental Toxicology",
"Chapter 4: Clinical Toxicology",
"Chapter 5: Regulatory Toxicology"
],
"Urban Geography": [
"Chapter 1: Basics of Urban Geography",
"Chapter 2: Urbanization Patterns",
"Chapter 3: Urban Land Use",
"Chapter 4: Urban Social Geography",
"Chapter 5: Urban Planning and Policy"
],
"Veterinary Medicine": [
"Chapter 1: Basics of Veterinary Medicine",
"Chapter 2: Animal Anatomy and Physiology",
"Chapter 3: Veterinary Pathology",
"Chapter 4: Veterinary Pharmacology",
"Chapter 5: Clinical Veterinary Practices"
],
"Wildlife Conservation": [
"Chapter 1: Basics of Wildlife Conservation",
"Chapter 2: Conservation Biology",
"Chapter 3: Habitat Management",
"Chapter 4: Endangered Species",
"Chapter 5: Conservation Strategies and Policies"
],
"Advanced Organic Chemistry": [
"Chapter 1: Structure and Bonding",
"Chapter 2: Reaction Mechanisms",
"Chapter 3: Stereochemistry",
"Chapter 4: Organic Synthesis",
"Chapter 5: Spectroscopy"
],
"Agricultural Economics": [
"Chapter 1: Basics of Agricultural Economics",
"Chapter 2: Farm Management",
"Chapter 3: Agricultural Markets and Prices",
"Chapter 4: Agricultural Policy",
"Chapter 5: International Trade in Agriculture"
],
"Artificial Neural Networks": [
"Chapter 1: Introduction to Neural Networks",
"Chapter 2: Feedforward Neural Networks",
"Chapter 3: Convolutional Neural Networks",
"Chapter 4: Recurrent Neural Networks",
"Chapter 5: Applications of Neural Networks"
],
"Bioinformatics": [
"Chapter 1: Basics of Bioinformatics",
"Chapter 2: Sequence Alignment",
"Chapter 3: Phylogenetic Analysis",
"Chapter 4: Structural Bioinformatics",
"Chapter 5: Genomics and Proteomics"
],
"Civil Engineering": [
"Chapter 1: Basics of Civil Engineering",
"Chapter 2: Structural Engineering",
"Chapter 3: Geotechnical Engineering",
"Chapter 4: Transportation Engineering",
"Chapter 5: Water Resources Engineering"
],
"Climate Science": [
"Chapter 1: Introduction to Climate Science",
"Chapter 2: Atmospheric Processes",
"Chapter 3: Climate Modeling",
"Chapter 4: Climate Change Impacts",
"Chapter 5: Mitigation and Adaptation Strategies"
],
"Cognitive Psychology": [
"Chapter 1: Basics of Cognitive Psychology",
"Chapter 2: Perception and Attention",
"Chapter 3: Memory and Learning",
"Chapter 4: Language and Thought",
"Chapter 5: Decision Making"
],
"Cybersecurity Risk Management": [
"Chapter 1: Basics of Cybersecurity",
"Chapter 2: Risk Assessment",
"Chapter 3: Risk Mitigation Strategies",
"Chapter 4: Incident Response",
"Chapter 5: Compliance and Governance"
],
"Database Systems": [
"Chapter 1: Introduction to Databases",
"Chapter 2: Relational Database Design",
"Chapter 3: SQL and Query Optimization",
"Chapter 4: NoSQL Databases",
"Chapter 5: Database Security"
],
"Developmental Biology": [
"Chapter 1: Basics of Developmental Biology",
"Chapter 2: Early Embryonic Development",
"Chapter 3: Organogenesis",
"Chapter 4: Stem Cells and Regeneration",
"Chapter 5: Evolutionary Developmental Biology"
],
"Environmental Chemistry": [
"Chapter 1: Introduction to Environmental Chemistry",
"Chapter 2: Chemical Processes in the Atmosphere",
"Chapter 3: Soil and Water Chemistry",
"Chapter 4: Environmental Toxicology",
"Chapter 5: Pollution Control and Remediation"
],
"Food Science and Technology": [
"Chapter 1: Basics of Food Science",
"Chapter 2: Food Chemistry",
"Chapter 3: Food Microbiology",
"Chapter 4: Food Processing Technologies",
"Chapter 5: Food Quality and Safety"
],
"Geology": [
"Chapter 1: Introduction to Geology",
"Chapter 2: Mineralogy",
"Chapter 3: Petrology",
"Chapter 4: Structural Geology",
"Chapter 5: Sedimentology and Stratigraphy"
],
"Healthcare Management": [
"Chapter 1: Basics of Healthcare Management",
"Chapter 2: Healthcare Systems and Policies",
"Chapter 3: Financial Management in Healthcare",
"Chapter 4: Quality Improvement in Healthcare",
"Chapter 5: Healthcare Information Systems"
],
"Human Resource Management": [
"Chapter 1: Introduction to HRM",
"Chapter 2: Recruitment and Selection",
"Chapter 3: Training and Development",
"Chapter 4: Performance Management",
"Chapter 5: Employee Relations"
],
"International Relations": [
"Chapter 1: Basics of International Relations",
"Chapter 2: Theories of International Relations",
"Chapter 3: International Organizations",
"Chapter 4: Global Security",
"Chapter 5: International Political Economy"
],
"Marine Biology": [
"Chapter 1: Introduction to Marine Biology",
"Chapter 2: Marine Ecosystems",
"Chapter 3: Marine Biodiversity",
"Chapter 4: Marine Physiology",
"Chapter 5: Conservation and Management"
],
"Microeconomics": [
"Chapter 1: Basics of Microeconomics",
"Chapter 2: Consumer Behavior",
"Chapter 3: Production and Costs",
"Chapter 4: Market Structures",
"Chapter 5: Market Failures and Public Policy"
],
"Nutrition Science": [
"Chapter 1: Basics of Nutrition",
"Chapter 2: Macronutrients and Micronutrients",
"Chapter 3: Nutrition Through Life Stages",
"Chapter 4: Diet and Chronic Diseases",
"Chapter 5: Public Health Nutrition"
],
"Optical Engineering": [
"Chapter 1: Basics of Optical Engineering",
"Chapter 2: Geometrical Optics",
"Chapter 3: Physical Optics",
"Chapter 4: Optical Instrumentation",
"Chapter 5: Laser Systems"
],
"Public Health": [
"Chapter 1: Basics of Public Health",
"Chapter 2: Epidemiology",
"Chapter 3: Environmental Health",
"Chapter 4: Health Policy and Management",
"Chapter 5: Global Health Issues"
],
"Robotics": [
"Chapter 1: Basics of Robotics",
"Chapter 2: Kinematics and Dynamics",
"Chapter 3: Sensors and Actuators",
"Chapter 4: Robot Control Systems",
"Chapter 5: Applications of Robotics"
],
"Social Work Practice": [
"Chapter 1: Basics of Social Work",
"Chapter 2: Social Work Theories and Methods",
"Chapter 3: Social Work with Individuals",
"Chapter 4: Social Work with Groups",
"Chapter 5: Community Practice"
],
"Soil Science": [
"Chapter 1: Basics of Soil Science",
"Chapter 2: Soil Formation and Classification",
"Chapter 3: Soil Physics",
"Chapter 4: Soil Chemistry",
"Chapter 5: Soil Fertility and Management"
],
"Systems Thinking": [
"Chapter 1: Introduction to Systems Thinking",
"Chapter 2: Systems Dynamics",
"Chapter 3: Feedback Loops",
"Chapter 4: Modeling and Simulation",
"Chapter 5: Applications in Problem Solving"
],
"Urban Studies": [
"Chapter 1: Basics of Urban Studies",
"Chapter 2: Urbanization and Urban Development",
"Chapter 3: Urban Sociology",
"Chapter 4: Urban Policy and Planning",
"Chapter 5: Sustainable Urban Development"
],
"Veterinary Surgery": [
"Chapter 1: Basics of Veterinary Surgery",
"Chapter 2: Surgical Techniques",
"Chapter 3: Anesthesia and Pain Management",
"Chapter 4: Soft Tissue Surgery",
"Chapter 5: Orthopedic Surgery"
],
"Wildlife Management": [
"Chapter 1: Basics of Wildlife Management",
"Chapter 2: Habitat Conservation",
"Chapter 3: Wildlife Population Dynamics",
"Chapter 4: Wildlife Policy and Law",
"Chapter 5: Case Studies in Wildlife Management"
],
"Biochemical Engineering": [
"Chapter 1: Introduction to Biochemical Engineering",
"Chapter 2: Enzyme Kinetics",
"Chapter 3: Bioreactor Design",
"Chapter 4: Downstream Processing",
"Chapter 5: Fermentation Technology",
"Chapter 6: Metabolic Engineering",
"Chapter 7: Bioseparation Techniques",
"Chapter 8: Bioprocess Control",
"Chapter 9: Biopharmaceutical Production",
"Chapter 10: Applications and Innovations"
],
"Advanced Plant Pathology": [
"Chapter 1: Introduction to Plant Pathology",
"Chapter 2: Pathogen Identification",
"Chapter 3: Disease Cycles",
"Chapter 4: Host-Pathogen Interactions",
"Chapter 5: Fungal Diseases",
"Chapter 6: Bacterial and Viral Diseases",
"Chapter 7: Nematode Diseases",
"Chapter 8: Disease Management Strategies",
"Chapter 9: Integrated Pest Management",
"Chapter 10: Advances in Plant Pathology"
],
"Aerospace Materials": [
"Chapter 1: Introduction to Aerospace Materials",
"Chapter 2: Metals and Alloys",
"Chapter 3: Composite Materials",
"Chapter 4: Polymers and Ceramics",
"Chapter 5: Material Properties",
"Chapter 6: Material Selection",
"Chapter 7: Failure Analysis",
"Chapter 8: Material Testing",
"Chapter 9: Advances in Aerospace Materials",
"Chapter 10: Future Trends"
],
"Algorithm Design and Analysis": [
"Chapter 1: Basics of Algorithm Design",
"Chapter 2: Divide and Conquer",
"Chapter 3: Dynamic Programming",
"Chapter 4: Greedy Algorithms",
"Chapter 5: Graph Algorithms",
"Chapter 6: String Algorithms",
"Chapter 7: NP-Completeness",
"Chapter 8: Approximation Algorithms",
"Chapter 9: Randomized Algorithms",
"Chapter 10: Advanced Topics"
],
"Bioacoustics": [
"Chapter 1: Introduction to Bioacoustics",
"Chapter 2: Sound Production in Animals",
"Chapter 3: Acoustic Communication",
"Chapter 4: Hearing Mechanisms",
"Chapter 5: Sound Propagation",
"Chapter 6: Bioacoustic Recording Techniques",
"Chapter 7: Data Analysis",
"Chapter 8: Behavioral Ecology",
"Chapter 9: Conservation Applications",
"Chapter 10: Future Directions"
],
"Building Information Modeling (BIM)": [
"Chapter 1: Introduction to BIM",
"Chapter 2: BIM Software Tools",
"Chapter 3: BIM Implementation",
"Chapter 4: 3D Modeling",
"Chapter 5: Clash Detection",
"Chapter 6: BIM for Construction Management",
"Chapter 7: BIM for Facilities Management",
"Chapter 8: Collaborative Workflows",
"Chapter 9: Legal and Contractual Issues",
"Chapter 10: Future Trends in BIM"
],
"Climate Engineering": [
"Chapter 1: Introduction to Climate Engineering",
"Chapter 2: Solar Radiation Management",
"Chapter 3: Carbon Dioxide Removal",
"Chapter 4: Geoengineering Technologies",
"Chapter 5: Climate Modeling",
"Chapter 6: Environmental Impacts",
"Chapter 7: Ethical and Social Considerations",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies",
"Chapter 10: Future Directions"
],
"Comparative Anatomy": [
"Chapter 1: Introduction to Comparative Anatomy",
"Chapter 2: Vertebrate Anatomy",
"Chapter 3: Invertebrate Anatomy",
"Chapter 4: Functional Morphology",
"Chapter 5: Evolution of Anatomical Structures",
"Chapter 6: Anatomical Adaptations",
"Chapter 7: Comparative Embryology",
"Chapter 8: Paleoanatomy",
"Chapter 9: Anatomical Techniques",
"Chapter 10: Future Trends in Comparative Anatomy"
],
"Criminal Profiling": [
"Chapter 1: Introduction to Criminal Profiling",
"Chapter 2: Psychological Theories",
"Chapter 3: Behavioral Evidence Analysis",
"Chapter 4: Crime Scene Analysis",
"Chapter 5: Offender Typologies",
"Chapter 6: Geographic Profiling",
"Chapter 7: Case Studies",
"Chapter 8: Legal and Ethical Issues",
"Chapter 9: Advances in Profiling Techniques",
"Chapter 10: Future Directions"
],
"Ecological Modeling": [
"Chapter 1: Introduction to Ecological Modeling",
"Chapter 2: Population Models",
"Chapter 3: Community Models",
"Chapter 4: Ecosystem Models",
"Chapter 5: Spatial Models",
"Chapter 6: Model Parameterization",
"Chapter 7: Model Validation",
"Chapter 8: Applications of Ecological Models",
"Chapter 9: Case Studies",
"Chapter 10: Future Trends"
],
"Energy Economics": [
"Chapter 1: Introduction to Energy Economics",
"Chapter 2: Energy Supply and Demand",
"Chapter 3: Energy Markets",
"Chapter 4: Energy Pricing",
"Chapter 5: Renewable Energy Economics",
"Chapter 6: Energy Policy",
"Chapter 7: Environmental Impacts",
"Chapter 8: Energy Security",
"Chapter 9: Case Studies",
"Chapter 10: Future Trends in Energy Economics"
],
"Environmental Toxicology": [
"Chapter 1: Introduction to Environmental Toxicology",
"Chapter 2: Toxicokinetics",
"Chapter 3: Toxicodynamics",
"Chapter 4: Ecotoxicology",
"Chapter 5: Chemical Toxicants",
"Chapter 6: Biological Toxicants",
"Chapter 7: Risk Assessment",
"Chapter 8: Environmental Monitoring",
"Chapter 9: Remediation Strategies",
"Chapter 10: Future Directions in Environmental Toxicology"
],
"Forensic Anthropology": [
"Chapter 1: Introduction to Forensic Anthropology",
"Chapter 2: Osteology",
"Chapter 3: Human Identification",
"Chapter 4: Skeletal Trauma Analysis",
"Chapter 5: Taphonomy",
"Chapter 6: Age and Sex Estimation",
"Chapter 7: Forensic Archaeology",
"Chapter 8: Case Studies",
"Chapter 9: Ethical Considerations",
"Chapter 10: Advances in Forensic Anthropology"
],
"Geochemistry": [
"Chapter 1: Introduction to Geochemistry",
"Chapter 2: Geochemical Cycles",
"Chapter 3: Isotope Geochemistry",
"Chapter 4: Organic Geochemistry",
"Chapter 5: Aqueous Geochemistry",
"Chapter 6: Mineral Geochemistry",
"Chapter 7: Environmental Geochemistry",
"Chapter 8: Analytical Techniques",
"Chapter 9: Case Studies",
"Chapter 10: Future Directions in Geochemistry"
],
"Human Factors Engineering": [
"Chapter 1: Introduction to Human Factors Engineering",
"Chapter 2: Ergonomics",
"Chapter 3: Cognitive Engineering",
"Chapter 4: Human-Computer Interaction",
"Chapter 5: Human Error",
"Chapter 6: Safety Engineering",
"Chapter 7: Usability Testing",
"Chapter 8: Case Studies",
"Chapter 9: Applications in Industry",
"Chapter 10: Future Trends in Human Factors"
],
"International Environmental Law": [
"Chapter 1: Introduction to International Environmental Law",
"Chapter 2: Principles of Environmental Law",
"Chapter 3: Environmental Treaties",
"Chapter 4: International Organizations",
"Chapter 5: Compliance and Enforcement",
"Chapter 6: Dispute Resolution",
"Chapter 7: Case Studies",
"Chapter 8: Regional Environmental Agreements",
"Chapter 9: Environmental Law in Practice",
"Chapter 10: Future Directions"
],
"Marine Geophysics": [
"Chapter 1: Introduction to Marine Geophysics",
"Chapter 2: Seafloor Mapping",
"Chapter 3: Plate Tectonics",
"Chapter 4: Marine Sediments",
"Chapter 5: Geophysical Survey Techniques",
"Chapter 6: Oceanic Crust and Mantle",
"Chapter 7: Hydrothermal Vents",
"Chapter 8: Submarine Volcanism",
"Chapter 9: Case Studies",
"Chapter 10: Advances in Marine Geophysics"
],
"Paleoclimatology": [
"Chapter 1: Introduction to Paleoclimatology",
"Chapter 2: Climate Proxies",
"Chapter 3: Ice Cores",
"Chapter 4: Tree Rings",
"Chapter 5: Marine Sediments",
"Chapter 6: Speleothems",
"Chapter 7: Paleoclimate Modeling",
"Chapter 8: Past Climate Events",
"Chapter 9: Human Impacts on Past Climates",
"Chapter 10: Future Directions"
],
"Psychopharmacology": [
"Chapter 1: Introduction to Psychopharmacology",
"Chapter 2: Neurotransmitters and Receptors",
"Chapter 3: Pharmacokinetics",
"Chapter 4: Pharmacodynamics",
"Chapter 5: Drug Classes",
"Chapter 6: Behavioral Effects",
"Chapter 7: Therapeutic Applications",
"Chapter 8: Drug Abuse and Dependence",
"Chapter 9: Clinical Trials",
"Chapter 10: Advances in Psychopharmacology"
],
"Quantum Information Science": [
"Chapter 1: Introduction to Quantum Information Science",
"Chapter 2: Quantum Bits and Quantum Gates",
"Chapter 3: Quantum Entanglement",
"Chapter 4: Quantum Algorithms",
"Chapter 5: Quantum Cryptography",
"Chapter 6: Quantum Error Correction",
"Chapter 7: Quantum Computing Hardware",
"Chapter 8: Quantum Communication",
"Chapter 9: Quantum Simulation",
"Chapter 10: Future Trends in Quantum Information"
],
"Sociolinguistics": [
"Chapter 1: Introduction to Sociolinguistics",
"Chapter 2: Language Variation",
"Chapter 3: Language and Identity",
"Chapter 4: Language and Gender",
"Chapter 5: Language and Power",
"Chapter 6: Language Contact",
"Chapter 7: Bilingualism and Multilingualism",
"Chapter 8: Sociolinguistic Methods",
"Chapter 9: Language Policy and Planning",
"Chapter 10: Future Directions in Sociolinguistics"
],
"Spacecraft Systems Engineering": [
"Chapter 1: Introduction to Spacecraft Systems",
"Chapter 2: Spacecraft Design Process",
"Chapter 3: Propulsion Systems",
"Chapter 4: Power Systems",
"Chapter 5: Thermal Control Systems",
"Chapter 6: Attitude Control Systems",
"Chapter 7: Communication Systems",
"Chapter 8: Structural Systems",
"Chapter 9: Integration and Testing",
"Chapter 10: Future Trends in Spacecraft Systems"
],
"Sports Psychology": [
"Chapter 1: Introduction to Sports Psychology",
"Chapter 2: Motivation in Sports",
"Chapter 3: Psychological Skills Training",
"Chapter 4: Team Dynamics",
"Chapter 5: Stress and Anxiety Management",
"Chapter 6: Goal Setting",
"Chapter 7: Injury and Rehabilitation",
"Chapter 8: Youth Sports Psychology",
"Chapter 9: Professional Athlete Mental Health",
"Chapter 10: Future Directions in Sports Psychology"
],
"Structural Engineering": [
"Chapter 1: Introduction to Structural Engineering",
"Chapter 2: Structural Analysis",
"Chapter 3: Steel Structures",
"Chapter 4: Concrete Structures",
"Chapter 5: Timber Structures",
"Chapter 6: Structural Dynamics",
"Chapter 7: Earthquake Engineering",
"Chapter 8: Bridge Engineering",
"Chapter 9: Structural Rehabilitation",
"Chapter 10: Future Trends in Structural Engineering"
],
"Sustainable Urban Planning": [
"Chapter 1: Introduction to Sustainable Urban Planning",
"Chapter 2: Urban Land Use Planning",
"Chapter 3: Transportation Planning",
"Chapter 4: Green Infrastructure",
"Chapter 5: Urban Design Principles",
"Chapter 6: Housing and Community Development",
"Chapter 7: Environmental Impact Assessment",
"Chapter 8: Policy and Governance",
"Chapter 9: Case Studies in Sustainable Cities",
"Chapter 10: Future Trends in Urban Planning"
],
"Theoretical Chemistry": [
"Chapter 1: Introduction to Theoretical Chemistry",
"Chapter 2: Quantum Chemistry",
"Chapter 3: Computational Methods",
"Chapter 4: Molecular Dynamics",
"Chapter 5: Statistical Mechanics",
"Chapter 6: Electronic Structure Theory",
"Chapter 7: Reaction Dynamics",
"Chapter 8: Spectroscopy and Theory",
"Chapter 9: Applications in Material Science",
"Chapter 10: Future Trends in Theoretical Chemistry"
],
"Urban Economics": [
"Chapter 1: Introduction to Urban Economics",
"Chapter 2: Urbanization and Economic Development",
"Chapter 3: Housing Markets",
"Chapter 4: Land Use and Zoning",
"Chapter 5: Transportation Economics",
"Chapter 6: Urban Public Finance",
"Chapter 7: Economic Impact of Urban Policies",
"Chapter 8: Urban Labor Markets",
"Chapter 9: Economic Inequality in Cities",
"Chapter 10: Future Trends in Urban Economics"
],
"Water Resource Policy": [
"Chapter 1: Introduction to Water Resource Policy",
"Chapter 2: Water Rights and Allocation",
"Chapter 3: Water Quality Management",
"Chapter 4: Integrated Water Resource Management",
"Chapter 5: Urban Water Management",
"Chapter 6: Agricultural Water Use",
"Chapter 7: Industrial Water Use",
"Chapter 8: International Water Policy",
"Chapter 9: Case Studies",
"Chapter 10: Future Trends in Water Policy"
],
"Advanced Botany": [
"Chapter 1: Introduction to Botany",
"Chapter 2: Plant Cell Biology",
"Chapter 3: Plant Genetics",
"Chapter 4: Photosynthesis and Respiration",
"Chapter 5: Plant Anatomy and Morphology",
"Chapter 6: Plant Physiology",
"Chapter 7: Plant Ecology",
"Chapter 8: Plant Pathology",
"Chapter 9: Economic Botany",
"Chapter 10: Advances in Botanical Research"
],
"Astronomical Instrumentation": [
"Chapter 1: Introduction to Astronomical Instruments",
"Chapter 2: Optical Telescopes",
"Chapter 3: Radio Telescopes",
"Chapter 4: Space-Based Observatories",
"Chapter 5: Spectroscopy",
"Chapter 6: Detectors and Sensors",
"Chapter 7: Adaptive Optics",
"Chapter 8: Data Acquisition and Analysis",
"Chapter 9: Instrument Calibration",
"Chapter 10: Future Technologies in Astronomy"
],
"Behavioral Neuroscience": [
"Chapter 1: Introduction to Behavioral Neuroscience",
"Chapter 2: Neuroanatomy",
"Chapter 3: Neurophysiology",
"Chapter 4: Neurochemistry",
"Chapter 5: Sensory Systems",
"Chapter 6: Motor Systems",
"Chapter 7: Learning and Memory",
"Chapter 8: Emotion and Behavior",
"Chapter 9: Neurological Disorders",
"Chapter 10: Advances in Neuroscience"
],
"Chemical Thermodynamics": [
"Chapter 1: Introduction to Thermodynamics",
"Chapter 2: Laws of Thermodynamics",
"Chapter 3: Thermodynamic Properties",
"Chapter 4: Phase Equilibria",
"Chapter 5: Chemical Equilibrium",
"Chapter 6: Thermodynamics of Solutions",
"Chapter 7: Statistical Thermodynamics",
"Chapter 8: Thermodynamics of Interfaces",
"Chapter 9: Non-equilibrium Thermodynamics",
"Chapter 10: Applications in Chemical Engineering"
],
"Climate Dynamics": [
"Chapter 1: Introduction to Climate Dynamics",
"Chapter 2: Atmospheric Circulation",
"Chapter 3: Ocean Circulation",
"Chapter 4: Energy Balance",
"Chapter 5: Climate Modeling",
"Chapter 6: Natural Climate Variability",
"Chapter 7: Anthropogenic Climate Change",
"Chapter 8: Paleoclimate Dynamics",
"Chapter 9: Climate Feedback Mechanisms",
"Chapter 10: Future Climate Projections"
],
"Comparative Politics": [
"Chapter 1: Introduction to Comparative Politics",
"Chapter 2: Political Systems and Regimes",
"Chapter 3: Electoral Systems and Voting Behavior",
"Chapter 4: Political Parties and Interest Groups",
"Chapter 5: Government Structures",
"Chapter 6: Public Policy and Administration",
"Chapter 7: Political Economy",
"Chapter 8: Social Movements and Revolutions",
"Chapter 9: Case Studies in Comparative Politics",
"Chapter 10: Trends in Comparative Political Analysis"
],
"Ecological Economics": [
"Chapter 1: Introduction to Ecological Economics",
"Chapter 2: Economic and Ecological Systems",
"Chapter 3: Valuing Ecosystem Services",
"Chapter 4: Sustainable Development",
"Chapter 5: Environmental Policy",
"Chapter 6: Resource Economics",
"Chapter 7: Climate Change Economics",
"Chapter 8: Ecological Footprint Analysis",
"Chapter 9: Case Studies in Ecological Economics",
"Chapter 10: Future Directions"
],
"Environmental Biotechnology": [
"Chapter 1: Introduction to Environmental Biotechnology",
"Chapter 2: Bioremediation Techniques",
"Chapter 3: Waste Treatment Technologies",
"Chapter 4: Bioenergy Production",
"Chapter 5: Microbial Ecology",
"Chapter 6: Genetically Modified Organisms in Environmental Management",
"Chapter 7: Environmental Genomics",
"Chapter 8: Environmental Monitoring",
"Chapter 9: Case Studies in Environmental Biotechnology",
"Chapter 10: Advances in Environmental Biotechnology"
],
"Experimental Physics": [
"Chapter 1: Introduction to Experimental Physics",
"Chapter 2: Measurement Techniques",
"Chapter 3: Data Analysis and Error Analysis",
"Chapter 4: Experimental Design",
"Chapter 5: Solid State Physics Experiments",
"Chapter 6: Nuclear and Particle Physics Experiments",
"Chapter 7: Optical and Laser Physics Experiments",
"Chapter 8: Quantum Mechanics Experiments",
"Chapter 9: Astrophysics Experiments",
"Chapter 10: Innovations in Experimental Physics"
],
"Hydraulic Engineering": [
"Chapter 1: Introduction to Hydraulic Engineering",
"Chapter 2: Fluid Mechanics",
"Chapter 3: Open Channel Flow",
"Chapter 4: Hydraulic Structures",
"Chapter 5: Water Distribution Systems",
"Chapter 6: Hydraulic Modeling",
"Chapter 7: Sediment Transport",
"Chapter 8: River Engineering",
"Chapter 9: Urban Hydrology",
"Chapter 10: Case Studies and Applications"
],
"Marine Chemistry": [
"Chapter 1: Introduction to Marine Chemistry",
"Chapter 2: Chemical Oceanography",
"Chapter 3: Seawater Composition",
"Chapter 4: Marine Biogeochemical Cycles",
"Chapter 5: Marine Organic Chemistry",
"Chapter 6: Trace Metals in the Marine Environment",
"Chapter 7: Marine Pollution",
"Chapter 8: Analytical Techniques in Marine Chemistry",
"Chapter 9: Case Studies in Marine Chemistry",
"Chapter 10: Advances in Marine Chemistry"
],
"Media Studies": [
"Chapter 1: Introduction to Media Studies",
"Chapter 2: Media Theories and Concepts",
"Chapter 3: Media Industries",
"Chapter 4: Media Audiences",
"Chapter 5: Digital Media",
"Chapter 6: Media and Society",
"Chapter 7: Media Policy and Regulation",
"Chapter 8: Media Production and Content Creation",
"Chapter 9: Case Studies in Media Studies",
"Chapter 10: Future Trends in Media"
],
"Nutritional Biochemistry": [
"Chapter 1: Introduction to Nutritional Biochemistry",
"Chapter 2: Macronutrients",
"Chapter 3: Micronutrients",
"Chapter 4: Metabolic Pathways",
"Chapter 5: Nutrition and Metabolism",
"Chapter 6: Nutrient-Gene Interactions",
"Chapter 7: Clinical Nutrition",
"Chapter 8: Nutritional Epidemiology",
"Chapter 9: Advances in Nutritional Biochemistry",
"Chapter 10: Future Directions"
],
"Optoelectronics": [
"Chapter 1: Introduction to Optoelectronics",
"Chapter 2: Semiconductor Physics",
"Chapter 3: Light Emitting Devices",
"Chapter 4: Photodetectors",
"Chapter 5: Lasers",
"Chapter 6: Optical Fibers",
"Chapter 7: Optoelectronic Integrated Circuits",
"Chapter 8: Solar Cells",
"Chapter 9: Applications in Communication and Sensing",
"Chapter 10: Future Trends in Optoelectronics"
],
"Planetary Science": [
"Chapter 1: Introduction to Planetary Science",
"Chapter 2: Solar System Formation",
"Chapter 3: Planetary Geology",
"Chapter 4: Planetary Atmospheres",
"Chapter 5: Icy Moons and Dwarf Planets",
"Chapter 6: Planetary Magnetospheres",
"Chapter 7: Exoplanets",
"Chapter 8: Astrobiology",
"Chapter 9: Planetary Exploration Missions",
"Chapter 10: Future Directions in Planetary Science"
],
"Proteomics": [
"Chapter 1: Introduction to Proteomics",
"Chapter 2: Protein Structure and Function",
"Chapter 3: Protein Extraction and Separation",
"Chapter 4: Mass Spectrometry in Proteomics",
"Chapter 5: Protein Identification",
"Chapter 6: Quantitative Proteomics",
"Chapter 7: Post-Translational Modifications",
"Chapter 8: Bioinformatics in Proteomics",
"Chapter 9: Applications of Proteomics",
"Chapter 10: Advances in Proteomics"
],
"Renewable Energy Engineering": [
"Chapter 1: Introduction to Renewable Energy",
"Chapter 2: Solar Power Systems",
"Chapter 3: Wind Energy Systems",
"Chapter 4: Biomass Energy Systems",
"Chapter 5: Geothermal Energy Systems",
"Chapter 6: Hydro Power Systems",
"Chapter 7: Energy Storage Technologies",
"Chapter 8: Grid Integration",
"Chapter 9: Economic and Policy Aspects",
"Chapter 10: Future Trends in Renewable Energy"
],
"Scientific Computing": [
"Chapter 1: Introduction to Scientific Computing",
"Chapter 2: Numerical Methods",
"Chapter 3: Linear Algebra in Computing",
"Chapter 4: Differential Equations",
"Chapter 5: Optimization Techniques",
"Chapter 6: Monte Carlo Methods",
"Chapter 7: Parallel Computing",
"Chapter 8: Scientific Visualization",
"Chapter 9: High-Performance Computing",
"Chapter 10: Applications and Case Studies"
],
"Sustainable Agriculture": [
"Chapter 1: Introduction to Sustainable Agriculture",
"Chapter 2: Soil Health and Fertility",
"Chapter 3: Integrated Pest Management",
"Chapter 4: Crop Rotation and Diversity",
"Chapter 5: Water Conservation",
"Chapter 6: Organic Farming",
"Chapter 7: Agroforestry",
"Chapter 8: Sustainable Livestock Management",
"Chapter 9: Policy and Economic Considerations",
"Chapter 10: Case Studies in Sustainable Agriculture"
],
"Theoretical Computer Science": [
"Chapter 1: Introduction to Theoretical Computer Science",
"Chapter 2: Automata Theory",
"Chapter 3: Formal Languages",
"Chapter 4: Computability",
"Chapter 5: Complexity Theory",
"Chapter 6: Cryptography",
"Chapter 7: Information Theory",
"Chapter 8: Quantum Computing Theory",
"Chapter 9: Algorithmic Game Theory",
"Chapter 10: Future Directions in Theoretical Computer Science"
],
"Urban Ecology": [
"Chapter 1: Introduction to Urban Ecology",
"Chapter 2: Urban Ecosystems",
"Chapter 3: Biodiversity in Urban Areas",
"Chapter 4: Green Infrastructure",
"Chapter 5: Urban Water Management",
"Chapter 6: Urban Climate and Air Quality",
"Chapter 7: Socio-Ecological Systems",
"Chapter 8: Urban Planning and Ecology",
"Chapter 9: Case Studies in Urban Ecology",
"Chapter 10: Future Directions in Urban Ecology"
],
"Veterinary Epidemiology": [
"Chapter 1: Introduction to Veterinary Epidemiology",
"Chapter 2: Disease Surveillance",
"Chapter 3: Disease Outbreak Investigation",
"Chapter 4: Epidemiological Methods",
"Chapter 5: Zoonotic Diseases",
"Chapter 6: Vaccination and Disease Control",
"Chapter 7: Risk Assessment",
"Chapter 8: Statistical Analysis in Epidemiology",
"Chapter 9: Case Studies in Veterinary Epidemiology",
"Chapter 10: Advances in Veterinary Epidemiology"
],
"Wildlife Ecology": [
"Chapter 1: Introduction to Wildlife Ecology",
"Chapter 2: Habitat Selection",
"Chapter 3: Population Dynamics",
"Chapter 4: Behavioral Ecology",
"Chapter 5: Predator-Prey Interactions",
"Chapter 6: Conservation Biology",
"Chapter 7: Wildlife Management",
"Chapter 8: Human-Wildlife Conflict",
"Chapter 9: Case Studies in Wildlife Ecology",
"Chapter 10: Future Directions in Wildlife Ecology"
],
"Zoology": [
"Chapter 1: Introduction to Zoology",
"Chapter 2: Invertebrate Zoology",
"Chapter 3: Vertebrate Zoology",
"Chapter 4: Animal Physiology",
"Chapter 5: Animal Behavior",
"Chapter 6: Evolution and Systematics",
"Chapter 7: Ecology and Conservation",
"Chapter 8: Comparative Anatomy",
"Chapter 9: Developmental Biology",
"Chapter 10: Advances in Zoological Research"
],
"Advanced Materials Science": [
"Chapter 1: Introduction to Materials Science",
"Chapter 2: Crystallography",
"Chapter 3: Phase Diagrams",
"Chapter 4: Mechanical Properties of Materials",
"Chapter 5: Thermal Properties of Materials",
"Chapter 6: Electrical Properties of Materials",
"Chapter 7: Optical Properties of Materials",
"Chapter 8: Magnetic Properties of Materials",
"Chapter 9: Composite Materials",
"Chapter 10: Nanomaterials"
],
"Aging and Society": [
"Chapter 1: Introduction to Aging and Society",
"Chapter 2: Biological Aspects of Aging",
"Chapter 3: Psychological Aspects of Aging",
"Chapter 4: Social Aspects of Aging",
"Chapter 5: Economic Aspects of Aging",
"Chapter 6: Aging and Health Care",
"Chapter 7: Aging and Policy",
"Chapter 8: Cultural Perspectives on Aging",
"Chapter 9: Gerontology",
"Chapter 10: Future Trends in Aging Studies"
],
"Biophysics": [
"Chapter 1: Introduction to Biophysics",
"Chapter 2: Molecular Biophysics",
"Chapter 3: Cellular Biophysics",
"Chapter 4: Biophysical Techniques",
"Chapter 5: Computational Biophysics",
"Chapter 6: Biophysics of Macromolecules",
"Chapter 7: Biomechanics",
"Chapter 8: Neurobiophysics",
"Chapter 9: Medical Biophysics",
"Chapter 10: Advances in Biophysics"
],
"Cognitive Robotics": [
"Chapter 1: Introduction to Cognitive Robotics",
"Chapter 2: Perception in Robots",
"Chapter 3: Learning and Adaptation",
"Chapter 4: Cognitive Architectures",
"Chapter 5: Human-Robot Interaction",
"Chapter 6: Autonomous Navigation",
"Chapter 7: Robot Decision Making",
"Chapter 8: Multi-Robot Systems",
"Chapter 9: Applications of Cognitive Robotics",
"Chapter 10: Future Trends in Cognitive Robotics"
],
"Cultural Heritage Management": [
"Chapter 1: Introduction to Cultural Heritage Management",
"Chapter 2: Heritage Conservation Principles",
"Chapter 3: Cultural Heritage and Law",
"Chapter 4: Heritage Tourism",
"Chapter 5: Museum Studies",
"Chapter 6: Digital Heritage",
"Chapter 7: Community Involvement",
"Chapter 8: Funding and Resources",
"Chapter 9: Case Studies in Heritage Management",
"Chapter 10: Future Directions in Cultural Heritage Management"
],
"Ecotoxicology": [
"Chapter 1: Introduction to Ecotoxicology",
"Chapter 2: Fate of Pollutants",
"Chapter 3: Bioaccumulation",
"Chapter 4: Toxicity Testing",
"Chapter 5: Ecotoxicological Models",
"Chapter 6: Risk Assessment",
"Chapter 7: Environmental Monitoring",
"Chapter 8: Case Studies in Ecotoxicology",
"Chapter 9: Remediation Techniques",
"Chapter 10: Advances in Ecotoxicology"
],
"Environmental Health": [
"Chapter 1: Introduction to Environmental Health",
"Chapter 2: Environmental Epidemiology",
"Chapter 3: Environmental Toxicology",
"Chapter 4: Occupational Health",
"Chapter 5: Air Quality and Health",
"Chapter 6: Water Quality and Health",
"Chapter 7: Waste Management",
"Chapter 8: Climate Change and Health",
"Chapter 9: Environmental Health Policy",
"Chapter 10: Future Directions in Environmental Health"
],
"Food Safety and Quality": [
"Chapter 1: Introduction to Food Safety",
"Chapter 2: Microbial Hazards",
"Chapter 3: Chemical Hazards",
"Chapter 4: Physical Hazards",
"Chapter 5: Foodborne Illnesses",
"Chapter 6: HACCP",
"Chapter 7: Food Quality Assurance",
"Chapter 8: Regulatory Framework",
"Chapter 9: Food Safety Management Systems",
"Chapter 10: Advances in Food Safety and Quality"
],
"Green Chemistry": [
"Chapter 1: Introduction to Green Chemistry",
"Chapter 2: Principles of Green Chemistry",
"Chapter 3: Sustainable Synthesis",
"Chapter 4: Green Catalysis",
"Chapter 5: Renewable Resources",
"Chapter 6: Green Solvents",
"Chapter 7: Waste Minimization",
"Chapter 8: Energy Efficiency",
"Chapter 9: Case Studies in Green Chemistry",
"Chapter 10: Future Directions in Green Chemistry"
],
"Hydrology and Water Resources": [
"Chapter 1: Introduction to Hydrology",
"Chapter 2: Hydrological Processes",
"Chapter 3: Surface Water Hydrology",
"Chapter 4: Groundwater Hydrology",
"Chapter 5: Hydrological Modeling",
"Chapter 6: Water Resources Management",
"Chapter 7: Water Quality",
"Chapter 8: Climate Change and Water Resources",
"Chapter 9: Case Studies in Hydrology",
"Chapter 10: Advances in Hydrological Science"
],
"Marine Ecology": [
"Chapter 1: Introduction to Marine Ecology",
"Chapter 2: Marine Ecosystems",
"Chapter 3: Marine Biodiversity",
"Chapter 4: Marine Food Webs",
"Chapter 5: Coral Reefs",
"Chapter 6: Mangroves and Seagrasses",
"Chapter 7: Marine Conservation",
"Chapter 8: Human Impacts on Marine Life",
"Chapter 9: Marine Protected Areas",
"Chapter 10: Climate Change and the Ocean"
],
"Neuroinformatics": [
"Chapter 1: Introduction to Neuroinformatics",
"Chapter 2: Brain Imaging Techniques",
"Chapter 3: Neurodata Management",
"Chapter 4: Computational Neuroscience",
"Chapter 5: Neuroinformatics Tools",
"Chapter 6: Data Analysis in Neuroscience",
"Chapter 7: Brain-Computer Interfaces",
"Chapter 8: Neuroinformatics Applications",
"Chapter 9: Ethical Issues in Neuroinformatics",
"Chapter 10: Future Directions in Neuroinformatics"
],
"Paleontology": [
"Chapter 1: Introduction to Paleontology",
"Chapter 2: Fossilization Processes",
"Chapter 3: Invertebrate Paleontology",
"Chapter 4: Vertebrate Paleontology",
"Chapter 5: Paleobotany",
"Chapter 6: Paleoecology",
"Chapter 7: Evolutionary Biology",
"Chapter 8: Paleontological Techniques",
"Chapter 9: Major Fossil Discoveries",
"Chapter 10: Future Directions in Paleontology"
],
"Philosophy of Science": [
"Chapter 1: Introduction to Philosophy of Science",
"Chapter 2: Scientific Method",
"Chapter 3: Theory and Observation",
"Chapter 4: Scientific Explanation",
"Chapter 5: Realism and Anti-Realism",
"Chapter 6: Science and Ethics",
"Chapter 7: Sociology of Science",
"Chapter 8: Philosophy of Biology",
"Chapter 9: Philosophy of Physics",
"Chapter 10: Contemporary Issues in Philosophy of Science"
],
"Sustainable Development": [
"Chapter 1: Introduction to Sustainable Development",
"Chapter 2: Environmental Sustainability",
"Chapter 3: Economic Sustainability",
"Chapter 4: Social Sustainability",
"Chapter 5: Sustainable Development Goals",
"Chapter 6: Policy and Governance",
"Chapter 7: Sustainable Urban Development",
"Chapter 8: Sustainable Agriculture",
"Chapter 9: Case Studies in Sustainable Development",
"Chapter 10: Future Trends in Sustainable Development"
],
"Systems Biology": [
"Chapter 1: Introduction to Systems Biology",
"Chapter 2: Biological Networks",
"Chapter 3: Mathematical Modeling",
"Chapter 4: Computational Methods",
"Chapter 5: Omics Technologies",
"Chapter 6: Data Integration",
"Chapter 7: Cellular Systems",
"Chapter 8: Organismal Systems",
"Chapter 9: Applications of Systems Biology",
"Chapter 10: Future Directions in Systems Biology"
],
"Tissue Engineering": [
"Chapter 1: Introduction to Tissue Engineering",
"Chapter 2: Cell Sources and Culture",
"Chapter 3: Scaffold Materials",
"Chapter 4: Bioreactors",
"Chapter 5: Tissue Regeneration",
"Chapter 6: Biomaterials",
"Chapter 7: Clinical Applications",
"Chapter 8: Regulatory and Ethical Issues",
"Chapter 9: Case Studies in Tissue Engineering",
"Chapter 10: Advances in Tissue Engineering"
],
"Virtual Reality Development": [
"Chapter 1: Introduction to Virtual Reality",
"Chapter 2: VR Hardware and Software",
"Chapter 3: 3D Modeling for VR",
"Chapter 4: VR Programming",
"Chapter 5: User Experience in VR",
"Chapter 6: VR Interaction Design",
"Chapter 7: VR Audio and Sound Design",
"Chapter 8: Performance Optimization",
"Chapter 9: VR Applications and Case Studies",
"Chapter 10: Future Trends in Virtual Reality"
],
"Water Management": [
"Chapter 1: Introduction to Water Management",
"Chapter 2: Water Resources Planning",
"Chapter 3: Water Distribution Systems",
"Chapter 4: Wastewater Management",
"Chapter 5: Stormwater Management",
"Chapter 6: Water Quality Monitoring",
"Chapter 7: Integrated Water Resource Management",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Water Management",
"Chapter 10: Future Trends in Water Management"
],
"Wildlife Rehabilitation": [
"Chapter 1: Introduction to Wildlife Rehabilitation",
"Chapter 2: Animal Rescue Techniques",
"Chapter 3: Medical Care for Wildlife",
"Chapter 4: Rehabilitation Protocols",
"Chapter 5: Release and Monitoring",
"Chapter 6: Wildlife Nutrition",
"Chapter 7: Behavioral Rehabilitation",
"Chapter 8: Legal and Ethical Issues",
"Chapter 9: Case Studies in Wildlife Rehabilitation",
"Chapter 10: Advances in Wildlife Rehabilitation"
],
"Zoo Management": [
"Chapter 1: Introduction to Zoo Management",
"Chapter 2: Animal Care and Welfare",
"Chapter 3: Exhibit Design and Maintenance",
"Chapter 4: Conservation Programs",
"Chapter 5: Education and Outreach",
"Chapter 6: Zoo Marketing and Fundraising",
"Chapter 7: Staff Training and Development",
"Chapter 8: Safety and Risk Management",
"Chapter 9: Case Studies in Zoo Management",
"Chapter 10: Future Trends in Zoo Management"
],
"Agricultural Biotechnology": [
"Chapter 1: Introduction to Agricultural Biotechnology",
"Chapter 2: Genetic Engineering in Crops",
"Chapter 3: Biotechnology in Animal Husbandry",
"Chapter 4: Biotechnology in Pest Management",
"Chapter 5: Biotechnology in Soil Health",
"Chapter 6: Biofertilizers and Biopesticides",
"Chapter 7: Regulatory Aspects",
"Chapter 8: Ethical Considerations",
"Chapter 9: Case Studies in Agricultural Biotechnology",
"Chapter 10: Future Trends in Agricultural Biotechnology"
],
"Art Therapy": [
"Chapter 1: Introduction to Art Therapy",
"Chapter 2: Theoretical Frameworks",
"Chapter 3: Techniques and Modalities",
"Chapter 4: Art Therapy with Children",
"Chapter 5: Art Therapy with Adults",
"Chapter 6: Art Therapy in Clinical Settings",
"Chapter 7: Art Therapy for Trauma",
"Chapter 8: Ethical and Legal Issues",
"Chapter 9: Case Studies in Art Therapy",
"Chapter 10: Advances in Art Therapy"
],
"Bioenergy": [
"Chapter 1: Introduction to Bioenergy",
"Chapter 2: Biomass Resources",
"Chapter 3: Biofuel Production Technologies",
"Chapter 4: Biogas and Biohydrogen",
"Chapter 5: Bioenergy Conversion Processes",
"Chapter 6: Environmental Impact of Bioenergy",
"Chapter 7: Economic Aspects of Bioenergy",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Bioenergy",
"Chapter 10: Future Trends in Bioenergy"
],
"Cardiovascular Physiology": [
"Chapter 1: Introduction to Cardiovascular Physiology",
"Chapter 2: Anatomy of the Heart",
"Chapter 3: Cardiac Electrophysiology",
"Chapter 4: Hemodynamics",
"Chapter 5: Regulation of Blood Flow",
"Chapter 6: Cardiac Cycle",
"Chapter 7: Pathophysiology of Cardiovascular Diseases",
"Chapter 8: Diagnostic Techniques",
"Chapter 9: Therapeutic Interventions",
"Chapter 10: Advances in Cardiovascular Research"
],
"Computational Linguistics": [
"Chapter 1: Introduction to Computational Linguistics",
"Chapter 2: Linguistic Data Processing",
"Chapter 3: Morphological Analysis",
"Chapter 4: Syntactic Parsing",
"Chapter 5: Semantic Analysis",
"Chapter 6: Machine Translation",
"Chapter 7: Speech Recognition and Synthesis",
"Chapter 8: Information Retrieval",
"Chapter 9: Applications in AI",
"Chapter 10: Future Directions in Computational Linguistics"
],
"Digital Humanities": [
"Chapter 1: Introduction to Digital Humanities",
"Chapter 2: Digital Archives and Libraries",
"Chapter 3: Text Encoding and Analysis",
"Chapter 4: Digital Mapping",
"Chapter 5: Data Visualization",
"Chapter 6: Digital Storytelling",
"Chapter 7: Digital Pedagogy",
"Chapter 8: Ethics in Digital Humanities",
"Chapter 9: Case Studies in Digital Humanities",
"Chapter 10: Future Trends in Digital Humanities"
],
"Ethnobotany": [
"Chapter 1: Introduction to Ethnobotany",
"Chapter 2: Traditional Plant Use",
"Chapter 3: Medicinal Plants",
"Chapter 4: Ethnobotanical Research Methods",
"Chapter 5: Cultural Significance of Plants",
"Chapter 6: Ethnobotany and Conservation",
"Chapter 7: Economic Botany",
"Chapter 8: Case Studies in Ethnobotany",
"Chapter 9: Ethnobotany in Modern Medicine",
"Chapter 10: Future Trends in Ethnobotany"
],
"Forest Ecology": [
"Chapter 1: Introduction to Forest Ecology",
"Chapter 2: Forest Ecosystems",
"Chapter 3: Forest Dynamics",
"Chapter 4: Forest Biodiversity",
"Chapter 5: Forest Soils",
"Chapter 6: Forest Hydrology",
"Chapter 7: Forest Conservation",
"Chapter 8: Human Impacts on Forests",
"Chapter 9: Forest Management",
"Chapter 10: Advances in Forest Ecology"
],
"Global Health": [
"Chapter 1: Introduction to Global Health",
"Chapter 2: Health Determinants",
"Chapter 3: Infectious Diseases",
"Chapter 4: Non-Communicable Diseases",
"Chapter 5: Global Health Systems",
"Chapter 6: Health Policy and Governance",
"Chapter 7: Global Health Ethics",
"Chapter 8: Case Studies in Global Health",
"Chapter 9: Health Interventions",
"Chapter 10: Future Trends in Global Health"
],
"Historical Linguistics": [
"Chapter 1: Introduction to Historical Linguistics",
"Chapter 2: Language Change",
"Chapter 3: Phonological Change",
"Chapter 4: Morphological Change",
"Chapter 5: Syntactic Change",
"Chapter 6: Semantic Change",
"Chapter 7: Comparative Method",
"Chapter 8: Language Families",
"Chapter 9: Language Reconstruction",
"Chapter 10: Advances in Historical Linguistics"
],
"Infectious Disease Epidemiology": [
"Chapter 1: Introduction to Infectious Disease Epidemiology",
"Chapter 2: Disease Transmission",
"Chapter 3: Outbreak Investigation",
"Chapter 4: Surveillance Systems",
"Chapter 5: Vaccination Strategies",
"Chapter 6: Antimicrobial Resistance",
"Chapter 7: Global Health Security",
"Chapter 8: Case Studies in Infectious Disease",
"Chapter 9: Disease Modeling",
"Chapter 10: Advances in Infectious Disease Epidemiology"
],
"Marine Resource Management": [
"Chapter 1: Introduction to Marine Resource Management",
"Chapter 2: Marine Ecosystem Services",
"Chapter 3: Fisheries Management",
"Chapter 4: Marine Protected Areas",
"Chapter 5: Marine Spatial Planning",
"Chapter 6: Sustainable Aquaculture",
"Chapter 7: Marine Pollution",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Marine Resource Management",
"Chapter 10: Advances in Marine Resource Management"
],
"Microbial Genetics": [
"Chapter 1: Introduction to Microbial Genetics",
"Chapter 2: Bacterial Genetics",
"Chapter 3: Viral Genetics",
"Chapter 4: Genetic Transfer Mechanisms",
"Chapter 5: Microbial Genomics",
"Chapter 6: Gene Regulation",
"Chapter 7: Genetic Engineering",
"Chapter 8: Applications in Biotechnology",
"Chapter 9: Case Studies in Microbial Genetics",
"Chapter 10: Advances in Microbial Genetics"
],
"Molecular Medicine": [
"Chapter 1: Introduction to Molecular Medicine",
"Chapter 2: Molecular Basis of Disease",
"Chapter 3: Genomic Medicine",
"Chapter 4: Molecular Diagnostics",
"Chapter 5: Targeted Therapies",
"Chapter 6: Gene Therapy",
"Chapter 7: Stem Cell Therapy",
"Chapter 8: Clinical Applications",
"Chapter 9: Case Studies in Molecular Medicine",
"Chapter 10: Advances in Molecular Medicine"
],
"Nanomaterials": [
"Chapter 1: Introduction to Nanomaterials",
"Chapter 2: Synthesis of Nanomaterials",
"Chapter 3: Characterization Techniques",
"Chapter 4: Properties of Nanomaterials",
"Chapter 5: Nanocomposites",
"Chapter 6: Applications in Electronics",
"Chapter 7: Applications in Medicine",
"Chapter 8: Environmental Applications",
"Chapter 9: Safety and Toxicology",
"Chapter 10: Future Trends in Nanomaterials"
],
"Pediatric Nursing": [
"Chapter 1: Introduction to Pediatric Nursing",
"Chapter 2: Growth and Development",
"Chapter 3: Pediatric Assessment",
"Chapter 4: Common Pediatric Illnesses",
"Chapter 5: Pediatric Emergency Care",
"Chapter 6: Chronic Conditions in Children",
"Chapter 7: Pediatric Pharmacology",
"Chapter 8: Family-Centered Care",
"Chapter 9: Ethical and Legal Issues",
"Chapter 10: Advances in Pediatric Nursing"
],
"Political Psychology": [
"Chapter 1: Introduction to Political Psychology",
"Chapter 2: Political Attitudes and Beliefs",
"Chapter 3: Voting Behavior",
"Chapter 4: Political Leadership",
"Chapter 5: Media and Politics",
"Chapter 6: Political Ideologies",
"Chapter 7: Group Behavior and Politics",
"Chapter 8: Public Opinion",
"Chapter 9: Case Studies in Political Psychology",
"Chapter 10: Future Directions in Political Psychology"
],
"Quantum Optics": [
"Chapter 1: Introduction to Quantum Optics",
"Chapter 2: Photons and Light",
"Chapter 3: Quantum States of Light",
"Chapter 4: Quantum Entanglement",
"Chapter 5: Quantum Measurement",
"Chapter 6: Quantum Communication",
"Chapter 7: Quantum Computing",
"Chapter 8: Experimental Techniques",
"Chapter 9: Applications of Quantum Optics",
"Chapter 10: Advances in Quantum Optics"
],
"Renewable Energy Policy": [
"Chapter 1: Introduction to Renewable Energy Policy",
"Chapter 2: Energy Policy Frameworks",
"Chapter 3: Incentives for Renewable Energy",
"Chapter 4: Regulatory Approaches",
"Chapter 5: International Energy Policy",
"Chapter 6: Policy Case Studies",
"Chapter 7: Socio-Economic Impacts",
"Chapter 8: Environmental Impacts",
"Chapter 9: Policy Implementation and Challenges",
"Chapter 10: Future Trends in Renewable Energy Policy"
],
"Science Communication": [
"Chapter 1: Introduction to Science Communication",
"Chapter 2: Principles of Effective Communication",
"Chapter 3: Communicating to Different Audiences",
"Chapter 4: Science Writing",
"Chapter 5: Digital and Social Media",
"Chapter 6: Public Engagement with Science",
"Chapter 7: Science in the Media",
"Chapter 8: Ethical Issues in Science Communication",
"Chapter 9: Case Studies in Science Communication",
"Chapter 10: Future Directions in Science Communication"
],
"Social Entrepreneurship": [
"Chapter 1: Introduction to Social Entrepreneurship",
"Chapter 2: Identifying Social Problems",
"Chapter 3: Business Models for Social Impact",
"Chapter 4: Financing Social Ventures",
"Chapter 5: Measuring Social Impact",
"Chapter 6: Scaling Social Enterprises",
"Chapter 7: Policy and Regulation",
"Chapter 8: Case Studies in Social Entrepreneurship",
"Chapter 9: Challenges in Social Entrepreneurship",
"Chapter 10: Future Trends in Social Entrepreneurship"
],
"Sociology of Education": [
"Chapter 1: Introduction to Sociology of Education",
"Chapter 2: Education and Socialization",
"Chapter 3: Educational Inequality",
"Chapter 4: Schooling and Society",
"Chapter 5: Education Policy",
"Chapter 6: Higher Education",
"Chapter 7: Education and Social Mobility",
"Chapter 8: Global Perspectives on Education",
"Chapter 9: Case Studies in Sociology of Education",
"Chapter 10: Future Directions in Sociology of Education"
],
"Structural Biology": [
"Chapter 1: Introduction to Structural Biology",
"Chapter 2: Protein Structure Determination",
"Chapter 3: Nucleic Acids Structures",
"Chapter 4: Membrane Proteins",
"Chapter 5: Structure-Function Relationships",
"Chapter 6: Structural Genomics",
"Chapter 7: Computational Structural Biology",
"Chapter 8: Techniques in Structural Biology",
"Chapter 9: Case Studies",
"Chapter 10: Advances in Structural Biology"
],
"Sustainable Tourism": [
"Chapter 1: Introduction to Sustainable Tourism",
"Chapter 2: Principles of Sustainable Tourism",
"Chapter 3: Ecotourism",
"Chapter 4: Cultural Heritage Tourism",
"Chapter 5: Community-Based Tourism",
"Chapter 6: Sustainable Tourism Practices",
"Chapter 7: Tourism Impact Assessment",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Sustainable Tourism",
"Chapter 10: Future Trends in Sustainable Tourism"
],
"Urban Sociology": [
"Chapter 1: Introduction to Urban Sociology",
"Chapter 2: Theories of Urbanization",
"Chapter 3: Urban Communities",
"Chapter 4: Urban Inequality",
"Chapter 5: Urban Politics",
"Chapter 6: Urban Culture",
"Chapter 7: Migration and Urbanization",
"Chapter 8: Housing and Homelessness",
"Chapter 9: Case Studies in Urban Sociology",
"Chapter 10: Future Directions in Urban Sociology"
],
"Veterinary Pharmacology": [
"Chapter 1: Introduction to Veterinary Pharmacology",
"Chapter 2: Drug Absorption and Distribution",
"Chapter 3: Pharmacokinetics",
"Chapter 4: Pharmacodynamics",
"Chapter 5: Veterinary Drug Classes",
"Chapter 6: Adverse Drug Reactions",
"Chapter 7: Veterinary Therapeutics",
"Chapter 8: Regulatory Aspects",
"Chapter 9: Case Studies in Veterinary Pharmacology",
"Chapter 10: Advances in Veterinary Pharmacology"
],
"Wildlife Conservation": [
"Chapter 1: Introduction to Wildlife Conservation",
"Chapter 2: Conservation Biology",
"Chapter 3: Habitat Conservation",
"Chapter 4: Species Conservation",
"Chapter 5: Conservation Genetics",
"Chapter 6: Conservation Policy",
"Chapter 7: Community-Based Conservation",
"Chapter 8: Wildlife Management",
"Chapter 9: Case Studies in Wildlife Conservation",
"Chapter 10: Future Directions in Wildlife Conservation"
],
"Advanced Manufacturing Technologies": [
"Chapter 1: Introduction to Advanced Manufacturing",
"Chapter 2: Additive Manufacturing",
"Chapter 3: Robotics in Manufacturing",
"Chapter 4: Advanced Materials Processing",
"Chapter 5: Smart Manufacturing",
"Chapter 6: Manufacturing Automation",
"Chapter 7: Quality Control in Manufacturing",
"Chapter 8: Sustainable Manufacturing",
"Chapter 9: Case Studies in Advanced Manufacturing",
"Chapter 10: Future Trends in Manufacturing Technologies"
],
"Anthropology of Religion": [
"Chapter 1: Introduction to Anthropology of Religion",
"Chapter 2: Theories of Religion",
"Chapter 3: Rituals and Beliefs",
"Chapter 4: Religious Practices",
"Chapter 5: Religion and Society",
"Chapter 6: Religious Change",
"Chapter 7: Comparative Religion",
"Chapter 8: Religion and Politics",
"Chapter 9: Case Studies in Anthropology of Religion",
"Chapter 10: Future Directions in Anthropology of Religion"
],
"Applied Econometrics": [
"Chapter 1: Introduction to Econometrics",
"Chapter 2: Regression Analysis",
"Chapter 3: Time Series Analysis",
"Chapter 4: Panel Data Analysis",
"Chapter 5: Instrumental Variables",
"Chapter 6: Econometric Models",
"Chapter 7: Hypothesis Testing",
"Chapter 8: Applications in Economics",
"Chapter 9: Case Studies in Applied Econometrics",
"Chapter 10: Advances in Econometric Techniques"
],
"Art and Technology": [
"Chapter 1: Introduction to Art and Technology",
"Chapter 2: Digital Art",
"Chapter 3: Interactive Media",
"Chapter 4: Virtual and Augmented Reality",
"Chapter 5: Art and Artificial Intelligence",
"Chapter 6: Art and Robotics",
"Chapter 7: Art and Biotechnology",
"Chapter 8: Case Studies in Art and Technology",
"Chapter 9: Ethical Issues",
"Chapter 10: Future Trends in Art and Technology"
],
"Biological Oceanography": [
"Chapter 1: Introduction to Biological Oceanography",
"Chapter 2: Marine Plankton",
"Chapter 3: Marine Food Webs",
"Chapter 4: Marine Benthos",
"Chapter 5: Marine Productivity",
"Chapter 6: Ocean Ecosystems",
"Chapter 7: Human Impacts on Marine Life",
"Chapter 8: Conservation of Marine Biodiversity",
"Chapter 9: Case Studies in Biological Oceanography",
"Chapter 10: Advances in Biological Oceanography"
],
"Biomaterials Science": [
"Chapter 1: Introduction to Biomaterials",
"Chapter 2: Properties of Biomaterials",
"Chapter 3: Synthesis and Characterization",
"Chapter 4: Biomaterials for Tissue Engineering",
"Chapter 5: Biomaterials in Drug Delivery",
"Chapter 6: Biocompatibility and Toxicity",
"Chapter 7: Applications in Medicine",
"Chapter 8: Regulatory and Ethical Issues",
"Chapter 9: Case Studies in Biomaterials",
"Chapter 10: Future Trends in Biomaterials Science"
],
"Biostatistics": [
"Chapter 1: Introduction to Biostatistics",
"Chapter 2: Descriptive Statistics",
"Chapter 3: Probability Theory",
"Chapter 4: Inferential Statistics",
"Chapter 5: Regression Analysis",
"Chapter 6: Survival Analysis",
"Chapter 7: Longitudinal Data Analysis",
"Chapter 8: Clinical Trials",
"Chapter 9: Case Studies in Biostatistics",
"Chapter 10: Advances in Biostatistical Methods"
],
"Business Ethics": [
"Chapter 1: Introduction to Business Ethics",
"Chapter 2: Ethical Theories and Principles",
"Chapter 3: Corporate Social Responsibility",
"Chapter 4: Ethical Decision Making",
"Chapter 5: Ethical Issues in Marketing",
"Chapter 6: Ethics in Human Resource Management",
"Chapter 7: Environmental Ethics",
"Chapter 8: Ethics in Global Business",
"Chapter 9: Case Studies in Business Ethics",
"Chapter 10: Future Directions in Business Ethics"
],
"Cognitive Science": [
"Chapter 1: Introduction to Cognitive Science",
"Chapter 2: Perception and Attention",
"Chapter 3: Memory and Learning",
"Chapter 4: Language and Thought",
"Chapter 5: Decision Making",
"Chapter 6: Cognitive Neuroscience",
"Chapter 7: Artificial Intelligence",
"Chapter 8: Computational Models of Cognition",
"Chapter 9: Case Studies in Cognitive Science",
"Chapter 10: Future Trends in Cognitive Science"
],
"Comparative Law": [
"Chapter 1: Introduction to Comparative Law",
"Chapter 2: Legal Traditions",
"Chapter 3: Civil Law Systems",
"Chapter 4: Common Law Systems",
"Chapter 5: Religious Law Systems",
"Chapter 6: Customary Law Systems",
"Chapter 7: Comparative Legal Analysis",
"Chapter 8: International Legal Harmonization",
"Chapter 9: Case Studies in Comparative Law",
"Chapter 10: Future Directions in Comparative Law"
],
"Computational Chemistry": [
"Chapter 1: Introduction to Computational Chemistry",
"Chapter 2: Quantum Chemistry Methods",
"Chapter 3: Molecular Dynamics",
"Chapter 4: Computational Thermodynamics",
"Chapter 5: Electronic Structure Calculations",
"Chapter 6: Computational Spectroscopy",
"Chapter 7: Drug Design and Discovery",
"Chapter 8: Materials Modeling",
"Chapter 9: Case Studies in Computational Chemistry",
"Chapter 10: Advances in Computational Chemistry"
],
"Cultural Anthropology": [
"Chapter 1: Introduction to Cultural Anthropology",
"Chapter 2: Culture and Society",
"Chapter 3: Kinship and Social Organization",
"Chapter 4: Religion and Belief Systems",
"Chapter 5: Economic Anthropology",
"Chapter 6: Political Anthropology",
"Chapter 7: Symbolism and Communication",
"Chapter 8: Anthropology of Art",
"Chapter 9: Case Studies in Cultural Anthropology",
"Chapter 10: Future Directions in Cultural Anthropology"
],
"Cybersecurity and Privacy": [
"Chapter 1: Introduction to Cybersecurity",
"Chapter 2: Threats and Vulnerabilities",
"Chapter 3: Cryptography",
"Chapter 4: Network Security",
"Chapter 5: Cybersecurity Policies",
"Chapter 6: Incident Response",
"Chapter 7: Privacy Principles",
"Chapter 8: Legal and Ethical Issues",
"Chapter 9: Case Studies in Cybersecurity",
"Chapter 10: Future Trends in Cybersecurity"
],
"Data Visualization": [
"Chapter 1: Introduction to Data Visualization",
"Chapter 2: Principles of Design",
"Chapter 3: Data Types and Sources",
"Chapter 4: Visualization Techniques",
"Chapter 5: Interactive Visualizations",
"Chapter 6: Visualization Tools and Software",
"Chapter 7: Storytelling with Data",
"Chapter 8: Evaluating Visualizations",
"Chapter 9: Case Studies in Data Visualization",
"Chapter 10: Future Trends in Data Visualization"
],
"Disaster Management": [
"Chapter 1: Introduction to Disaster Management",
"Chapter 2: Types of Disasters",
"Chapter 3: Risk Assessment and Mitigation",
"Chapter 4: Disaster Preparedness",
"Chapter 5: Emergency Response",
"Chapter 6: Disaster Recovery",
"Chapter 7: Community Resilience",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Disaster Management",
"Chapter 10: Advances in Disaster Management"
],
"Ecological Restoration": [
"Chapter 1: Introduction to Ecological Restoration",
"Chapter 2: Principles of Restoration Ecology",
"Chapter 3: Restoration Techniques",
"Chapter 4: Soil and Water Restoration",
"Chapter 5: Native Species Reintroduction",
"Chapter 6: Monitoring and Evaluation",
"Chapter 7: Social and Cultural Aspects",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in Ecological Restoration",
"Chapter 10: Future Trends in Restoration Ecology"
],
"Educational Technology": [
"Chapter 1: Introduction to Educational Technology",
"Chapter 2: Learning Theories and Technology",
"Chapter 3: Instructional Design",
"Chapter 4: E-Learning and Online Education",
"Chapter 5: Educational Software and Tools",
"Chapter 6: Multimedia in Education",
"Chapter 7: Mobile Learning",
"Chapter 8: Evaluation and Assessment",
"Chapter 9: Case Studies in Educational Technology",
"Chapter 10: Future Trends in Educational Technology"
],
"Environmental Geology": [
"Chapter 1: Introduction to Environmental Geology",
"Chapter 2: Earth Materials and Processes",
"Chapter 3: Natural Hazards",
"Chapter 4: Soil and Water Resources",
"Chapter 5: Environmental Impact of Mining",
"Chapter 6: Waste Management",
"Chapter 7: Land Use Planning",
"Chapter 8: Environmental Policy and Regulation",
"Chapter 9: Case Studies in Environmental Geology",
"Chapter 10: Advances in Environmental Geology"
],
"Food Microbiology": [
"Chapter 1: Introduction to Food Microbiology",
"Chapter 2: Microorganisms in Food",
"Chapter 3: Food Spoilage",
"Chapter 4: Foodborne Pathogens",
"Chapter 5: Fermentation and Food Production",
"Chapter 6: Microbial Testing in Food Safety",
"Chapter 7: Probiotics and Prebiotics",
"Chapter 8: Biotechnology in Food Microbiology",
"Chapter 9: Case Studies in Food Microbiology",
"Chapter 10: Advances in Food Microbiology"
],
"Forensic Psychology": [
"Chapter 1: Introduction to Forensic Psychology",
"Chapter 2: Criminal Behavior",
"Chapter 3: Psychological Assessment",
"Chapter 4: Expert Testimony",
"Chapter 5: Offender Rehabilitation",
"Chapter 6: Victimology",
"Chapter 7: Juvenile Justice",
"Chapter 8: Legal and Ethical Issues",
"Chapter 9: Case Studies in Forensic Psychology",
"Chapter 10: Advances in Forensic Psychology"
],
"Genetic Counseling": [
"Chapter 1: Introduction to Genetic Counseling",
"Chapter 2: Principles of Genetics",
"Chapter 3: Genetic Testing and Diagnosis",
"Chapter 4: Counseling Techniques",
"Chapter 5: Ethical Issues in Genetic Counseling",
"Chapter 6: Psychosocial Aspects",
"Chapter 7: Prenatal Genetic Counseling",
"Chapter 8: Cancer Genetic Counseling",
"Chapter 9: Case Studies in Genetic Counseling",
"Chapter 10: Advances in Genetic Counseling"
],
"Historical Preservation": [
"Chapter 1: Introduction to Historical Preservation",
"Chapter 2: Preservation Techniques",
"Chapter 3: Historical Research Methods",
"Chapter 4: Legal and Ethical Issues",
"Chapter 5: Conservation of Built Heritage",
"Chapter 6: Cultural Landscape Preservation",
"Chapter 7: Digital Preservation",
"Chapter 8: Community Involvement",
"Chapter 9: Case Studies in Historical Preservation",
"Chapter 10: Future Trends in Historical Preservation"
],
"Human Computer Interaction": [
"Chapter 1: Introduction to Human-Computer Interaction",
"Chapter 2: User-Centered Design",
"Chapter 3: Usability Testing",
"Chapter 4: Interaction Styles",
"Chapter 5: Accessibility and Inclusive Design",
"Chapter 6: Mobile Interaction",
"Chapter 7: Social and Ethical Issues",
"Chapter 8: Advanced HCI Topics",
"Chapter 9: Case Studies in HCI",
"Chapter 10: Future Trends in Human-Computer Interaction"
],
"Immunology": [
"Chapter 1: Introduction to Immunology",
"Chapter 2: Components of the Immune System",
"Chapter 3: Innate Immunity",
"Chapter 4: Adaptive Immunity",
"Chapter 5: Immunological Disorders",
"Chapter 6: Immunotherapy",
"Chapter 7: Vaccines and Vaccination",
"Chapter 8: Immunological Techniques",
"Chapter 9: Case Studies in Immunology",
"Chapter 10: Advances in Immunology"
],
"Intelligent Transportation Systems": [
"Chapter 1: Introduction to Intelligent Transportation Systems",
"Chapter 2: Traffic Management Systems",
"Chapter 3: Public Transportation Systems",
"Chapter 4: Autonomous Vehicles",
"Chapter 5: ITS Technologies",
"Chapter 6: Data Collection and Analysis",
"Chapter 7: Safety and Security",
"Chapter 8: Policy and Regulation",
"Chapter 9: Case Studies in ITS",
"Chapter 10: Future Trends in Intelligent Transportation Systems"
],
"Marine Biotechnology": [
"Chapter 1: Introduction to Marine Biotechnology",
"Chapter 2: Marine Bioresources",
"Chapter 3: Marine Bioproducts",
"Chapter 4: Marine Bioprocessing",
"Chapter 5: Aquaculture Biotechnology",
"Chapter 6: Environmental Biotechnology",
"Chapter 7: Marine Pharmaceuticals",
"Chapter 8: Regulatory and Ethical Issues",
"Chapter 9: Case Studies in Marine Biotechnology",
"Chapter 10: Advances in Marine Biotechnology"
],
"Mathematical Modeling": [
"Chapter 1: Introduction to Mathematical Modeling",
"Chapter 2: Modeling Techniques",
"Chapter 3: Linear Models",
"Chapter 4: Nonlinear Models",
"Chapter 5: Dynamical Systems",
"Chapter 6: Stochastic Models",
"Chapter 7: Applications in Biology",
"Chapter 8: Applications in Economics",
"Chapter 9: Case Studies in Mathematical Modeling",
"Chapter 10: Advances in Mathematical Modeling"
],
"Molecular Diagnostics": [
"Chapter 1: Introduction to Molecular Diagnostics",
"Chapter 2: DNA and RNA Analysis",
"Chapter 3: Polymerase Chain Reaction",
"Chapter 4: Next-Generation Sequencing",
"Chapter 5: Microarray Technology",
"Chapter 6: Diagnostic Biomarkers",
"Chapter 7: Clinical Applications",
"Chapter 8: Regulatory and Ethical Issues",
"Chapter 9: Case Studies in Molecular Diagnostics",
"Chapter 10: Advances in Molecular Diagnostics"
],
"Music Therapy": [
"Chapter 1: Introduction to Music Therapy",
"Chapter 2: Theoretical Foundations",
"Chapter 3: Techniques and Interventions",
"Chapter 4: Music Therapy with Children",
"Chapter 5: Music Therapy with Adults",
"Chapter 6: Music Therapy in Clinical Settings",
"Chapter 7: Research Methods in Music Therapy",
"Chapter 8: Ethical and Legal Issues",
"Chapter 9: Case Studies in Music Therapy",
"Chapter 10: Advances in Music Therapy"
],
"Nanomedicine": [
"Chapter 1: Introduction to Nanomedicine",
"Chapter 2: Nanoparticles in Medicine",
"Chapter 3: Drug Delivery Systems",
"Chapter 4: Diagnostic Nanotechnologies",
"Chapter 5: Therapeutic Nanotechnologies",
"Chapter 6: Regulatory and Ethical Issues",
"Chapter 7: Clinical Applications",
"Chapter 8: Research Methods",
"Chapter 9: Case Studies in Nanomedicine",
"Chapter 10: Future Trends in Nanomedicine"
],
"Natural Language Processing": [
"Chapter 1: Introduction to Natural Language Processing",
"Chapter 2: Text Preprocessing",
"Chapter 3: Language Modeling",
"Chapter 4: Text Classification",
"Chapter 5: Named Entity Recognition",
"Chapter 6: Part-of-Speech Tagging",
"Chapter 7: Syntax and Parsing",
"Chapter 8: Sentiment Analysis",
"Chapter 9: Machine Translation",
"Chapter 10: Information Retrieval",
"Chapter 11: Question Answering",
"Chapter 12: Text Summarization",
"Chapter 13: Dialogue Systems",
"Chapter 14: Ethical Issues in NLP",
"Chapter 15: Advances in Natural Language Processing"
],
"Paleoclimatology": [
"Chapter 1: Introduction to Paleoclimatology",
"Chapter 2: Climate Archives",
"Chapter 3: Ice Cores",
"Chapter 4: Sediment Cores",
"Chapter 5: Tree Rings",
"Chapter 6: Coral Records",
"Chapter 7: Paleoclimate Modeling",
"Chapter 8: Quaternary Climate Change",
"Chapter 9: Proxy Data Analysis",
"Chapter 10: Paleoclimate and Human Evolution",
"Chapter 11: Climate Forcing Mechanisms",
"Chapter 12: Past Climate Events",
"Chapter 13: Paleoclimate and Future Projections",
"Chapter 14: Field Methods in Paleoclimatology",
"Chapter 15: Advances in Paleoclimatology"
],
"Pharmacogenomics": [
"Chapter 1: Introduction to Pharmacogenomics",
"Chapter 2: Genetic Variation and Drug Response",
"Chapter 3: Pharmacokinetics and Pharmacodynamics",
"Chapter 4: Genotyping Technologies",
"Chapter 5: Pharmacogenomic Biomarkers",
"Chapter 6: Personalized Medicine",
"Chapter 7: Clinical Implementation",
"Chapter 8: Ethical and Legal Issues",
"Chapter 9: Case Studies in Pharmacogenomics",
"Chapter 10: Pharmacogenomics in Oncology",
"Chapter 11: Pharmacogenomics in Psychiatry",
"Chapter 12: Drug Development and Pharmacogenomics",
"Chapter 13: Future Trends in Pharmacogenomics",
"Chapter 14: Research Methods in Pharmacogenomics",
"Chapter 15: Advances in Pharmacogenomics"
],
"Quantum Computing": [
"Chapter 1: Introduction to Quantum Computing",
"Chapter 2: Qubits and Quantum Gates",
"Chapter 3: Quantum Algorithms",
"Chapter 4: Quantum Entanglement",
"Chapter 5: Quantum Error Correction",
"Chapter 6: Quantum Cryptography",
"Chapter 7: Quantum Machine Learning",
"Chapter 8: Quantum Hardware",
"Chapter 9: Quantum Simulation",
"Chapter 10: Quantum Computing Languages",
"Chapter 11: Quantum Computing Applications",
"Chapter 12: Challenges in Quantum Computing",
"Chapter 13: Quantum Computing Research",
"Chapter 14: Ethical Implications",
"Chapter 15: Future Directions in Quantum Computing"
],
"Regenerative Medicine": [
"Chapter 1: Introduction to Regenerative Medicine",
"Chapter 2: Stem Cells",
"Chapter 3: Tissue Engineering",
"Chapter 4: Biomaterials in Regeneration",
"Chapter 5: Cell Therapy",
"Chapter 6: Gene Therapy",
"Chapter 7: Immunotherapy",
"Chapter 8: Clinical Applications",
"Chapter 9: Regulatory and Ethical Issues",
"Chapter 10: Research Methods",
"Chapter 11: Advances in Regenerative Medicine",
"Chapter 12: Challenges in Regenerative Medicine",
"Chapter 13: Case Studies",
"Chapter 14: Future Trends in Regenerative Medicine",
"Chapter 15: Integration with Traditional Therapies"
],
"Robotics Engineering": [
"Chapter 1: Introduction to Robotics Engineering",
"Chapter 2: Kinematics",
"Chapter 3: Dynamics",
"Chapter 4: Control Systems",
"Chapter 5: Sensors and Actuators",
"Chapter 6: Robot Programming",
"Chapter 7: Autonomous Robots",
"Chapter 8: Mobile Robots",
"Chapter 9: Industrial Robotics",
"Chapter 10: Robotics in Medicine",
"Chapter 11: Human-Robot Interaction",
"Chapter 12: Ethical and Legal Issues",
"Chapter 13: Research in Robotics",
"Chapter 14: Case Studies in Robotics",
"Chapter 15: Future Trends in Robotics Engineering"
],
"Sociocultural Anthropology": [
"Chapter 1: Introduction to Sociocultural Anthropology",
"Chapter 2: Theories of Culture",
"Chapter 3: Ethnographic Methods",
"Chapter 4: Kinship and Family",
"Chapter 5: Religion and Ritual",
"Chapter 6: Political Systems",
"Chapter 7: Economic Systems",
"Chapter 8: Language and Communication",
"Chapter 9: Gender and Sexuality",
"Chapter 10: Globalization",
"Chapter 11: Cultural Change",
"Chapter 12: Health and Illness",
"Chapter 13: Case Studies in Sociocultural Anthropology",
"Chapter 14: Ethical Issues",
"Chapter 15: Future Directions in Sociocultural Anthropology"
],
"Space Weather": [
"Chapter 1: Introduction to Space Weather",
"Chapter 2: The Sun and Solar Wind",
"Chapter 3: Earth's Magnetosphere",
"Chapter 4: Geomagnetic Storms",
"Chapter 5: Ionospheric Disturbances",
"Chapter 6: Space Weather Forecasting",
"Chapter 7: Impact on Satellites",
"Chapter 8: Impact on Communications",
"Chapter 9: Impact on Power Systems",
"Chapter 10: Space Weather and Aviation",
"Chapter 11: Space Weather Observatories",
"Chapter 12: Case Studies in Space Weather",
"Chapter 13: Research Methods",
"Chapter 14: Advances in Space Weather Prediction",
"Chapter 15: Future Trends in Space Weather"
],
"Sustainable Architecture": [
"Chapter 1: Introduction to Sustainable Architecture",
"Chapter 2: Principles of Sustainable Design",
"Chapter 3: Energy Efficiency",
"Chapter 4: Sustainable Building Materials",
"Chapter 5: Passive Solar Design",
"Chapter 6: Water Conservation",
"Chapter 7: Indoor Environmental Quality",
"Chapter 8: Sustainable Urban Design",
"Chapter 9: Green Building Certification",
"Chapter 10: Case Studies in Sustainable Architecture",
"Chapter 11: Retrofitting Existing Buildings",
"Chapter 12: Policy and Regulation",
"Chapter 13: Research Methods",
"Chapter 14: Advances in Sustainable Building Technologies",
"Chapter 15: Future Trends in Sustainable Architecture"
],
"Synthetic Biology": [
"Chapter 1: Introduction to Synthetic Biology",
"Chapter 2: Genetic Circuits",
"Chapter 3: DNA Synthesis",
"Chapter 4: Metabolic Engineering",
"Chapter 5: Protein Engineering",
"Chapter 6: Synthetic Genomes",
"Chapter 7: Applications in Medicine",
"Chapter 8: Applications in Agriculture",
"Chapter 9: Ethical and Social Issues",
"Chapter 10: Regulatory Aspects",
"Chapter 11: Research Methods",
"Chapter 12: Case Studies",
"Chapter 13: Advances in Synthetic Biology",
"Chapter 14: Challenges in Synthetic Biology",
"Chapter 15: Future Directions in Synthetic Biology"
],
"Telemedicine": [
"Chapter 1: Introduction to Telemedicine",
"Chapter 2: Telemedicine Technologies",
"Chapter 3: Telehealth Applications",
"Chapter 4: Remote Patient Monitoring",
"Chapter 5: Telemedicine in Chronic Disease Management",
"Chapter 6: Legal and Ethical Issues",
"Chapter 7: Telemedicine in Mental Health",
"Chapter 8: Telemedicine in Rural Health",
"Chapter 9: Case Studies in Telemedicine",
"Chapter 10: Telemedicine Policy and Regulation",
"Chapter 11: Research Methods",
"Chapter 12: Advances in Telemedicine",
"Chapter 13: Challenges in Telemedicine",
"Chapter 14: Future Trends in Telemedicine",
"Chapter 15: Integration with Traditional Healthcare Systems"
],
"Urban Planning and Policy": [
"Chapter 1: Introduction to Urban Planning",
"Chapter 2: History of Urban Planning",
"Chapter 3: Land Use Planning",
"Chapter 4: Transportation Planning",
"Chapter 5: Housing Policy",
"Chapter 6: Environmental Planning",
"Chapter 7: Economic Development Planning",
"Chapter 8: Urban Design Principles",
"Chapter 9: Public Participation in Planning",
"Chapter 10: Case Studies in Urban Planning",
"Chapter 11: Planning for Resilience",
"Chapter 12: Policy and Regulation",
"Chapter 13: Research Methods",
"Chapter 14: Advances in Urban Planning",
"Chapter 15: Future Directions in Urban Planning"
],
"Bioinformatics": [
"Chapter 1: Introduction to Bioinformatics",
"Chapter 2: Biological Databases",
"Chapter 3: Sequence Alignment",
"Chapter 4: Genomic Data Analysis",
"Chapter 5: Protein Structure Prediction",
"Chapter 6: Phylogenetics",
"Chapter 7: Systems Biology",
"Chapter 8: Transcriptomics",
"Chapter 9: Metabolomics",
"Chapter 10: Data Integration",
"Chapter 11: Bioinformatics Tools and Software",
"Chapter 12: Case Studies in Bioinformatics",
"Chapter 13: Ethical Issues in Bioinformatics",
"Chapter 14: Research Methods",
"Chapter 15: Future Trends in Bioinformatics"
],
"Climate Science": [
"Chapter 1: Introduction to Climate Science",
"Chapter 2: Climate Systems",
"Chapter 3: Atmospheric Processes",
"Chapter 4: Oceanography",
"Chapter 5: Paleoclimatology",
"Chapter 6: Climate Change Evidence",
"Chapter 7: Climate Modeling",
"Chapter 8: Impacts of Climate Change",
"Chapter 9: Mitigation Strategies",
"Chapter 10: Adaptation Strategies",
"Chapter 11: Policy and Regulation",
"Chapter 12: Case Studies in Climate Science",
"Chapter 13: Ethical Issues in Climate Science",
"Chapter 14: Research Methods",
"Chapter 15: Future Trends in Climate Science"
],
"Cyber Law": [
"Chapter 1: Introduction to Cyber Law",
"Chapter 2: Legal Framework for Cybersecurity",
"Chapter 3: Data Protection and Privacy",
"Chapter 4: Intellectual Property in Cyberspace",
"Chapter 5: Cybercrime and Law Enforcement",
"Chapter 6: E-commerce and Online Contracts",
"Chapter 7: Social Media and the Law",
"Chapter 8: Ethical Issues in Cyber Law",
"Chapter 9: Case Studies in Cyber Law",
"Chapter 10: International Cyber Law",
"Chapter 11: Regulatory Compliance",
"Chapter 12: Research Methods",
"Chapter 13: Future Directions in Cyber Law",
"Chapter 14: Dispute Resolution in Cyberspace",
"Chapter 15: Advances in Cyber Law"
],
"Developmental Psychology": [
"Chapter 1: Introduction to Developmental Psychology",
"Chapter 2: Prenatal Development",
"Chapter 3: Infancy and Toddlerhood",
"Chapter 4: Early Childhood Development",
"Chapter 5: Middle Childhood Development",
"Chapter 6: Adolescence",
"Chapter 7: Adulthood and Aging",
"Chapter 8: Cognitive Development",
"Chapter 9: Social and Emotional Development",
"Chapter 10: Language Development",
"Chapter 11: Personality Development",
"Chapter 12: Research Methods in Developmental Psychology",
"Chapter 13: Case Studies",
"Chapter 14: Ethical Issues",
"Chapter 15: Future Directions in Developmental Psychology"
],
"Ecology": [
"Chapter 1: Introduction to Ecology",
"Chapter 2: Population Ecology",
"Chapter 3: Community Ecology",
"Chapter 4: Ecosystem Ecology",
"Chapter 5: Behavioral Ecology",
"Chapter 6: Evolutionary Ecology",
"Chapter 7: Conservation Ecology",
"Chapter 8: Urban Ecology",
"Chapter 9: Global Ecology",
"Chapter 10: Climate Change and Ecology",
"Chapter 11: Research Methods in Ecology",
"Chapter 12: Case Studies in Ecology",
"Chapter 13: Ethical Issues",
"Chapter 14: Advances in Ecology",
"Chapter 15: Future Trends in Ecology"
],
"Environmental Economics": [
"Chapter 1: Introduction to Environmental Economics",
"Chapter 2: Economic Principles",
"Chapter 3: Environmental Valuation",
"Chapter 4: Cost-Benefit Analysis",
"Chapter 5: Market Failure and Externalities",
"Chapter 6: Environmental Policy Instruments",
"Chapter 7: Natural Resource Economics",
"Chapter 8: Climate Change Economics",
"Chapter 9: Sustainable Development",
"Chapter 10: International Environmental Agreements",
"Chapter 11: Case Studies in Environmental Economics",
"Chapter 12: Research Methods",
"Chapter 13: Ethical Issues",
"Chapter 14: Advances in Environmental Economics",
"Chapter 15: Future Trends in Environmental Economics"
],
"Ethical Hacking": [
"Chapter 1: Introduction to Ethical Hacking",
"Chapter 2: Hacking Methodologies",
"Chapter 3: Network Security",
"Chapter 4: Vulnerability Assessment",
"Chapter 5: Penetration Testing",
"Chapter 6: Web Application Security",
"Chapter 7: Wireless Network Security",
"Chapter 8: Social Engineering",
"Chapter 9: Ethical Issues in Hacking",
"Chapter 10: Case Studies in Ethical Hacking",
"Chapter 11: Legal Aspects",
"Chapter 12: Tools and Techniques",
"Chapter 13: Research Methods",
"Chapter 14: Advances in Ethical Hacking",
"Chapter 15: Future Trends in Cybersecurity"
],
"Genomics": [
"Chapter 1: Introduction to Genomics",
"Chapter 2: Genome Sequencing",
"Chapter 3: Genome Annotation",
"Chapter 4: Functional Genomics",
"Chapter 5: Comparative Genomics",
"Chapter 6: Structural Genomics",
"Chapter 7: Epigenomics",
"Chapter 8: Transcriptomics",
"Chapter 9: Metagenomics",
"Chapter 10: Single-cell Genomics",
"Chapter 11: Bioinformatics Tools",
"Chapter 12: Case Studies in Genomics",
"Chapter 13: Ethical Issues",
"Chapter 14: Advances in Genomics",
"Chapter 15: Future Trends in Genomics"
],
"Hydrogeology": [
"Chapter 1: Introduction to Hydrogeology",
"Chapter 2: Groundwater Flow",
"Chapter 3: Aquifer Properties",
"Chapter 4: Groundwater Recharge",
"Chapter 5: Groundwater Contamination",
"Chapter 6: Groundwater Modeling",
"Chapter 7: Water Well Design",
"Chapter 8: Groundwater Management",
"Chapter 9: Groundwater and Climate Change",
"Chapter 10: Hydrogeological Field Methods",
"Chapter 11: Policy and Regulation",
"Chapter 12: Case Studies in Hydrogeology",
"Chapter 13: Ethical Issues",
"Chapter 14: Advances in Hydrogeology",
"Chapter 15: Future Trends in Hydrogeology"
],
"Information Systems Security": [
"Chapter 1: Introduction to Information Systems Security",
"Chapter 2: Security Policies and Procedures",
"Chapter 3: Risk Management",
"Chapter 4: Network Security",
"Chapter 5: Cryptography",
"Chapter 6: Application Security",
"Chapter 7: Cloud Security",
"Chapter 8: Identity and Access Management",
"Chapter 9: Incident Response",
"Chapter 10: Compliance and Legal Issues",
"Chapter 11: Case Studies in Information Systems Security",
"Chapter 12: Tools and Techniques",
"Chapter 13: Research Methods",
"Chapter 14: Advances in Information Systems Security",
"Chapter 15: Future Trends in Information Systems Security"
],
"Marine Geology": [
"Chapter 1: Introduction to Marine Geology",
"Chapter 2: Ocean Basin Formation",
"Chapter 3: Marine Sediments",
"Chapter 4: Plate Tectonics and Oceanic Crust",
"Chapter 5: Hydrothermal Vents",
"Chapter 6: Marine Geohazards",
"Chapter 7: Coastal Processes",
"Chapter 8: Sea-Level Change",
"Chapter 9: Marine Mineral Resources",
"Chapter 10: Paleoceanography",
"Chapter 11: Research Methods in Marine Geology",
"Chapter 12: Case Studies",
"Chapter 13: Ethical Issues",
"Chapter 14: Advances in Marine Geology",
"Chapter 15: Future Trends in Marine Geology"
],
"Metabolic Engineering": [
"Chapter 1: Introduction to Metabolic Engineering",
"Chapter 2: Cellular Metabolism",
"Chapter 3: Metabolic Pathway Design",
"Chapter 4: Genetic Engineering Techniques",
"Chapter 5: Metabolic Flux Analysis",
"Chapter 6: Synthetic Biology Approaches",
"Chapter 7: Applications in Biotechnology",
"Chapter 8: Biofuels and Biochemicals",
"Chapter 9: Metabolic Engineering for Pharmaceuticals",
"Chapter 10: Regulatory and Ethical Issues",
"Chapter 11: Case Studies",
"Chapter 12: Research Methods",
"Chapter 13: Advances in Metabolic Engineering",
"Chapter 14: Challenges in Metabolic Engineering",
"Chapter 15: Future Directions"
],
}
# Create a DataFrame with Topic and Chapters
data = []
for topic, chapters in courses_with_chapters.items():
combined_chapters = "\n".join(chapters)
data.append([topic, combined_chapters])
df = pd.DataFrame(data, columns=["topic", "response"])
print(df.shape)
</code>
<code>
# Fungsi untuk membuat teks pada kolom 'prompt'
def create_prompt(row):
chapters = row['response'].split('\n')
course_length = len(chapters)
course_title = row['topic']
prompt = f"List {course_length} chapter name for a course '{course_title}'.\n Write just the outline name only! no need for explanation Write just the outline name only! no need for explanation For example: Chapter 1: ___ Chapter 2: ___ Chapter 3: ___"
return prompt
# Membuat kolom baru 'prompt'
df['prompt'] = df.apply(create_prompt, axis=1)
# Mengubah urutan kolom
df = df[['topic', 'prompt', 'response']]
</code>
<code>
df
</code>
# Push to Hub
## Data Prep for Train
<code>
# Function to create the desired JSON structure
def create_sharegpt_style(row):
return [
{"from": "human", "value": row["prompt"]},
{"from": "gpt", "value": row["response"]}
]
# Apply the function to create the new column
df["sharegpt_style"] = df.apply(create_sharegpt_style, axis=1)
</code>
<code>
df
</code>
<code>
df.to_json("../../dataset/generate-chapter-dataset.jsonl", orient='records', lines=True)
</code>
<code>
from huggingface_hub import login
from datasets import Dataset
login()
dataset = Dataset.from_pandas(df)
dataset = dataset.train_test_split(test_size=0.2)
dataset.push_to_hub("fauzanrrizky/generate-chapters-dataset")
</code>
|
{
"filename": "generate_chapter_dataset_1.ipynb",
"repository": "AFF-Learntelligence/machine-learning",
"query": "transformed_from_existing",
"size": 355506,
"sha": ""
}
|
# bdn2022.ipynb
Repository: muzaale/denotas
```
#colleenhoover
— wtf is she?
— has no. 6, 8, 11, and 15 on Amazon
```
.
```
#modusoperandi
Ambition/Beyond
Morality/Good
Nyege Nyege/Evil
🏔
```
.
```
Dear GTPCI Advisory Committee members,
I have discussed the issue regarding my mentorship plan with Dr. Segev and the Department of Surgery. We do not expect a substantial impact on my GTPCI plans for the following reasons:
1. Dr. Segev will be keeping an adjust appointment with the JHU Department of Surgery;
2. For the last two years we’ve mostly communicated via zoom and email and will continue to do so on a weekly basis;
3. Our research group at JHU will continue close collaborations with the Dorry’s research group at NYU;
4. I will continue to have access to the data sources required for my research topic.
Because the GTPCI PhD is an integral part of my K08 career development plans, we have also addressed this issue with the National Institute of Aging. I’ve attached a copy of the letter for your convenience.
```
.
Soros, Paulson, Simons, Cohen, Ackman, Icahn, Graham
.
```
Icahn: The Restless Billionaire
30:11/1:42:06
“It’s almost unreadable, it’s so complex, but he got his revenge — and he won that senior thesis.”
```
.
```
#emotion
From emotional problems of the student
1.
2.Withers & dries in the healthy upperclassman or graduate
3.Lush Brazilian jungle of teenage fantasy (persists in neurosis)
#kindred
William James
James finally earned his MD degree in June 1869 but he never practiced medicine. What he called his "soul-sickness" would only be resolved in 1872, after an extended period of philosophical searching. He married Alice Gibbens in 1878. In 1882 he joined the Theosophical Society
"I originally studied medicine in order to be a physiologist, but I drifted into psychology and philosophy from a sort of fatality. I never had any philosophic instruction, the first lecture on psychology I ever heard being the first I ever gave"
```
.
```
#beer
https://www.beeradvocate.com/beer/profile/207/510204/
4.43/5 rDev +5.7%
look: 4.25 | smell: 4.25 | taste: 4.5 | feel: 4.5 | overall: 4.5
by BEERchitect from Kentucky
Never ones to rush out with new offerings, the brewery of Rochefort now finds it time to roll out a standout beer among their darker regular offering in the style of Tripel Ale. Nearly seventy years in the making, this lighter colored beer still packs that signature Rochefort punch.
Triple Extra pours with an elegant weave into the the classic Rochefort chalice while billowing to the brim with a cottony, stark-white froth. As odd as seeing a lighter hued beverage sitting in that glass, the scent is both exciting and unique as a collection of vinous spices, candied fruit, fresh dough and peppery hops drift onto the senses with a radiant but piquant perfume. Sweeter though in its early impressions, those fresh bread flavors lace with impressions of sugar cookie and honeysuckle.
As its expressive carbonation pops on the middle palate, the bulk of sweetness is lifted and the higher fruit impressions start to shine. Pear, apple, lemon, orange peel and a hint of grapefruit pull from the ester profile and invite a later balance of peppercorn, white wine and grassy hops to catch up to the level of fruitiness for a dry and even-keel character heading into finish.
Medium bodied and trending lighter and drier on the finish, the Tripel Ale is a masterful spice-forward and drier example of the style and done so in that classic Trappist Tripel style. Finishing surprisingly refreshing and effortless on the palate for its strength, the ale extends with an afterglow of yeast spice and peppery hops for a hint of lemongrass and white wine to close.
Thursday at 02:01 PM
```
.
```
#destiny
Only from my time and after me will politics on a large scale exits on earth
```
.
```
#jeen-yuhs
I can tell when a nigga is hot and when he has the potential to become complacent — Pharrell
Window seat video — Erykah Badu
College drop out, through the wire… perspective from a convalescent #whyamisowise?
```
.
```
#faves
In the mountains the shortest way is from peak to peak, but for that route thou must have long legs. Proverbs should be peaks, and those spoken to should be big and tall
I no longer feel in common with you; the very cloud which I see beneath me, the blackness and heaviness at which I laugh—that is your thunder-cloud
🏔
And to me also, who appreciate life, the butterflies, and soap-bubbles, and whatever is like them amongst us, seem most to enjoy happiness.
To see these light, foolish, pretty, lively little sprites flit about—that moveth Zarathustra to tears and songs
🏔
I should only believe in a God that would know how to dance.
And when I saw my devil, I found him serious, thorough, profound, solemn: he was the spirit of gravity—through him all things fall.
Not by wrath, but by laughter, do we slay. Come, let us slay the spirit of gravity!
```
.
```
#poe
Muzaale, Abimereki poe/phd/gtpci
https://jhjhm.zoom.us/j/98180375399
jhoover2@jhmi.edu
dorry@jhmi.edu
kbandee1@jhu.edu
dcelent1@jhu.edu
ermiller@jhmi.edu
fjohnst4@jhmi.edu
bcaffo1@jhu.edu
cdenard1@jhu.edu
```
.
```
#ken
— Kenny
— Victoria
— Savanna
— Jada
— Tyra
```
.
```
#philosophy
Robert Sapolsky’s on categorical thinking, origin of error
Evolution from Ape to man is not categorical, no “civilized”
The first nine aphorisms in Human, All Too Human
Cause and effect in Nietzsche’s writings on 02/28/22
Missing data lecture by Elizabeth Sugar on 03/01/22
And thoughts of Constantine Frangakis also on 03/01/22
Wittes et all dilemma as the DSMB of WHI studies of several exposures and outcomes
A complexity which is thousandfold reaches our consciousness as a simple entity — Nietzsche, 1888
No aphorism is more frequently repeated in connection with field trials, than that we must ask Nature few questions, or ideally one question, at a time. The writer is convinced that this view is wholly mistaken. Nature he suggests, will best respond to a logical and carefully thought out questionnaire.”
— R.A. Fisher, 1926
I develop designs and methods of analyses to evaluate treatments in medicine, public health and policy (causal inference) — Frangakis, 2002
*
Association between increased sophistication of analyses and number of assumptions made?
Randomized trials are not exempt given factor design, multiple outcomes, adverse effects, follow-up t
I mistrust all systematizers and avoid them. The will to a system is a lack of integrity — Nietzsche
*
I have never pondered over questions that are not questions. I have never squandered my strength. Of actual religious difficulties, for instance, I have no experience. I have never known what it feels to be “sinful”
*
To conclude, we value creation more than searching; engineering more than science; practical more than theoretical
#explainedvarianceorcauseandeffect
```
.
```
#love
The state in which man sees things most decidedly as they are not
```
#### 03/2022
```
#cheese
Reypenaer*
1. Nietszche
2. Socrates
3. Epicurus*
Loses 25% of weight during its historical ripening process (2.5-3 jaars)
x
1 jaar 200g
VSOP 200g
XO 580g
x
The Philosophy of Luxury. — A garden, figs, a little cheese, and three or four good friends — that was the luxury of Epicurus
x
Mozzarella
Toasted baguette 🥖
Tomato 🍅
Roasted peppers 🌶
```
.
```
#bourdain
Bourdain was known for his sarcastic comments about vegan and vegetarian activists, considering their lifestyle "rude" to the inhabitants of many countries he visited. He considered vegetarianism, except in the case of religious exemptions, a "First World luxury". However, he also believed that Americans eat too much meat, and admired vegetarians and vegans who put aside their beliefs when visiting different cultures in order to be respectful of their hosts
```
.
```
#patton
Through a Glass Darkly,
Perhaps I stabbed our SaviorIn His sacred helpless side.Yet I’ve called His name in blessingWhen in after times I died.
Through the travail of the agesMidst the pomp and toil of warHave I fought and strove and perishedCountless times upon this star.
I have sinned and I have sufferedPlayed the hero and the knaveFought for belly, shame or countryAnd for each have found a grave.
So as through a glass and darklyThe age long strife I seeWhere I fought in many guises,Many names – but always me.
So forever in the futureShall I battle as of yore,Dying to be born a fighterBut to die again once more.
https://spotterup.com/poetry-george-s-patton/
```
.
```
#9mileruns
11/13/18-3/25/22
Total 145
<8’30” 9 (93rd percentile)
=8’30” 9 (87th percentile)
&8’40” is a “B”
```
.
```
#willsmith
In his 432-page book published in November, called “Will,” Smith confided that for a time he would sometimes vomit after orgasming.
https://nypost.com/2022/03/28/will-smiths-wild-year-sex-reveals-to-crazy-oscars-outburst/
```
.
https://www.youtube.com/watch?v=rkfFyELXdoM
.
```
#thiel
5:40/22:19
https://www.youtube.com/watch?v=H5NUv0nOQCU
The Education Good:
— Investment: future
— Consumption: party
— Insurance: safety-net
— Tournament: zerosum
https://www.youtube.com/watch?v=WOEsVjqoOfA
2:40/19:37
```
.
```
#run
3/30/22
Last meal at 15:40
— Burger 🍔
— Fries 🍟
— Cheese Panini 🥪🧀
— Jalapeño grits
— Ketchup
```
.
```
Slide 1: Title
I’m going talk to you about the..
So.. A key part of donor evaluation is the estimation of the donor candidates risks, communicating these to the donor candidate, and reaching a shared decision on whether its appropriate to proceed with donation
Slide 2: Frank
Now.. A knowledge gap exists in our understanding of the risks faced by older donors
Here we have Frank, the oldest living donor to date – 84yo (Linda — 72yo neighbor). The informed consent process in this case could not have been based on actual data from older donors (i.e., maybe extrapolations from younger donors)
Slide 3: Data
To address this knowledge gap, I’ll be using national registry data to enumerate all the older donors in the US
Slide 4: Analysis
We can thus quantify the risks attributable to donation… the kind of information that is currently lacking for older donors.. and to be shared during the informed consent process
Slide 5: Challenges
Preceding outcomes that affect quality of life
No Gerontological syndromes not captured by ICD-10 codes..
Our findings will inform the conversation between candidate and provider about the propriety of donation
*NIS
Slide 6: Goals
These ideas translate into the following specific research aims: Our efforts will culminate in the creation of online risk calculators that inform the conversation between candidates and providers about the propriety of donation. Dr. Muzaale will learn how sentinel hospitalization events in an aging cohort of older donors leave a footprint of present and missing
Aim 1 & 2: info for medical community
Aim 3: more accessible info for donor candidates
Slide 7: Online-Calculators
A.
Nonparametric hazard of the base-case; maximum-likelihood estimate of the difference (on a log scale) between the hazard of the base-case and the hazard for the specific-case with explanatory variables X, and t-years (using base-case absolute risks and specific-case relative risk coefficients from the multivariable regression in Aims 1 and 2)
B.
In a manner analogous to what I just described for risk of ESRD/death, we will use multivariable logistic regression to describe nephrectomy attributable hospitalizations, associated medical diagnoses. Beta– maximum likelihood estimate of the additive effect on the log odds for a unit change in a given explanatory variable X
X
1.IRB, logistics
— acceleratomoerr
— hearing aids
—
Consenting, acquisition
— older subgroup.. 75
— server size
—EMR data l, lazy
— Big a problem? EModification
What do we know
How common is donation >50
— power and comments
— think about what to do if not power
Confounding by indication — in frailty
```
.
```
1.Julie
— Insurance companies & others may use your calculators for harm
— Consider IRB implications of proposed EMR: consenting, aquisition
— Publishing a paper that reduces access to donation
— Accelerometer and hearing aids: pertinent to gerontology and in NHANES/EMR
2.Karen
— Big picture (increasing trends for older donors)
— Gerontologist hat (confounding by indication in frailty for EMR: cause-inference methods)
— Statistics hat
— Effect-modification a big problem? What do we know? How common is donation over 50?
3.Pete
— Validating EMR for donors as NHANES has already be done
— Quality of EMR data: I’m a lazy clinician for instance
— Power size calculation: what difference in risk between groups is clinically relevant?
4.Celentano
—
5.Dorry
— Missing data mechanisms
— Validating new national registry
— OPTN oversight encouraging on clinical practice (e.g. eGFR in donors, CMS flags, etc)
```
.
```
#hitmen
1.Chucky Thompson
2.Stevie J
3.Mario Winans
```
.
#### 04/2022
```
#exam
Muzaale, POE/GTPCI PhD
04/01/22
1.Logical thinking
2.Breadth of knowledge
3.Develop and conduct research
10-15min presentation
```
.
```
1.Julie
— Insurance companies & others may use your calculators for harm
— Consider IRB implications of proposed EMR: consenting, aquisition
— Publishing a paper that reduces access to donation
— Accelerometer and hearing aids: pertinent to gerontology and in NHANES/EMR
2.Karen
— Big picture (increasing trends for older donors)
— Gerontologist hat (confounding by indication in frailty for EMR: cause-inference methods)
— Statistics hat
— Effect-modification a big problem? What do we know? How common is donation over 50?
3.Pete
— Validating EMR for donors as NHANES has already be done
— Quality of EMR data: I’m a lazy clinician for instance
— Power size calculation: what difference in risk between groups is clinically relevant?
4.Celentano
—
5.Dorry
— Missing data mechanisms
— Validating new national registry
— OPTN oversight encouraging on clinical practice (e.g. eGFR in donors, CMS flags, etc)
```
.
`#graçias``
04/01/22
10am-12pm
🙏🏾
.
```
#husbandsandwives
Roger/Ruth (Knows dad, tribalism)
Mutabazi/Brandy
Tayebwa/Anita, Joannita
Simeon/Shannie
```
.
```
#film
"I got out there to entertain audiences the way Hollywood entertained me. Make them forget their lives for a few hours and be thrilled, awed, or excited at the big room where make believe lives," Jackson continued
```
.
[Donald Lawrence](https://www.youtube.com/watch?v=kglSBsGCJWY)
.
```
#Morgann
1.MPH vs PhD? She liked my recommendation
2.From an affluent black family
3.West Baltimore
4.Transition in career
5.From federal government (Forestry)
6.And to health (MPH vs PhD nursing)
```
.
https://www.youtube.com/watch?v=4p4Cs2IVSXw
.
```
#death
To die proudly when it is no longer possible to live proudly. Death freely chosen, death at the right time, brightly and cheerfully accomplished amid children and witnesses: then a real farewell is still possible..
#36. Morality for physicians. Skirmishes of an untimely man. Twilight of Idols
If it was a dog you’d put it down
11:47min left, Afterlife E1:S1
(Here we are introduced to Tony’s cantankerous funk — which he calls a superpower — that is founded on a contradiction: an atheistic view of the afterlife, but an idealization of a wife lost to cancer)
Why do we give dogs a better death than we give ourselves?
https://aeon.co/essays/why-do-we-give-dogs-a-better-death-than-we-give-ourselves
```
.
```
#goingsoft
Had a girlfriend at every time between 2007-2018
Underestimated just how soft I’d grown in the interim
One needs a stern philosophy from as earlier in life as possible
```
.
```
#faves
Julius Caesar 15
Cesare Borgia 7
Napoleon 61
```
.
```
#borgias
This monk, with all the vengeful instincts of an unsuccessful priest in him, raised a rebellion against the Renaissance in Rome.... Instead of grasping, with profound thanksgiving, the miracle that had taken place: the conquest of Christianity at its capital — instead of this, his hatred was stimulated by the spectacle.
A religious man thinks only of himself. — Luther saw only the depravity of the papacy at the very moment when the opposite was becoming apparent: the old corruption, the peccatum originale, Christianity itself, no longer occupied the papal chair!
Instead there was life! Instead there was the triumph of life! Instead there was a great yea to all lofty, beautiful and daring things!... And Luther restored the church: he attacked it.... The Renaissance — an event without meaning, a great futility! — Ah, these Germans, what they have not cost us!
Futility — that has always been the work of the Germans. — The Reformation; Leibnitz; Kant and so-called German philosophy; the war of “liberation”; the empire — every time a futile substitute for something that once existed, for something irrecoverable.... These Germans, I confess, are my enemies: I despise all their uncleanliness in concept and valuation, their cowardice before every honest[…]”
```
.
```
#saidi
04/08/22
86min 07sec/17km
86.12 min/10.563 miles
8.15393/MI
8’09”/MI
Vs
74min 46sec/9miles
8’18”/MI
```
.
https://nypost.com/2022/04/07/will-smith-warns-jada-dont-use-me-for-clout-in-video/amp/
.
```
#football
Man City vs Liverpool
10:30am
Sunday 04/10/22
```
.
177.2 after swimming
178.9 after a Pilsner & Larger (anticipated)
.
```
#max’s
Max’s taphouse
There’s 3 Jason’s
Jason Saturday’s :-)
Jason 1 y’already know
Jason 2 no affect, really nice, deep eyes
Jason 3 slightly bald, big beard, shy, little affect, really really nice
Bob y’already know
Scooter (wife is with Bloomberg school)
Jeff (long-ass beard 🧔) confirmed
Diana/Big “D” (Cinco de Mayo birthday)
Jason (another one, who knows me, bushy 🧔, little affect!!)
Tim (young, capped 🧢 dude)
? Cheerful bold guy who knows me!
Mac: young, shy, goatee, cap, knows my name!!!!
??? Bold dude, knows me, somewhat cheerful, is NOT scooter :-) Jammie
Jammie is def the most cheerful 👨🦲:-)
Jay: knows me, no smiles, but really nice
Racheal: new 2021 chic
Didi: extremely pleasant big black dude. Upgraded to bartender from bouncer during the 2020 shitshow
Scooter really is the bold dude with
A wife at Hopkins who
Travels a lot to Africa and places
But who is the young dude??
Confirmed (by 🛴, whose wife actual works at JHPIEGO, but does some mentor-mentee shit at Bloomberg); the young dude with a reverse cap 🧢 is Tim (what are the odds???)
Jason — looks like there’s 3 of them
Yeah — 200% emotionless, no beard too
That’s also Jason! Jay!!
Rachael — hot Jewish
Matthew — young, southern accent (Nice guy)
```
.
```
#summary
Ambitious
L’homme moyen
Slacker
*
Mark 8: 34-38 : For what shall it profit a man, if he shall gain the whole world, and lose his own soul?
*
Examples of slacker protagonist becoming ambitious (aka losing their soul)
1.Henry V
2.William James
3.Not “The Dude”!
4.Cédric
5.[XXX]
“To those who want to know where the hell’s my ambition, I say: and you, where the hell’s your well-being? Usually they stammer a few words and I understand by them that they’ve postponed it. Because first, they need:
1.Money (and a job);
2.Husband/wife;
3.Car;
4.A house; and,
5.Retirement plan
And people say I’m the one wasting my time!
https://ir.lib.uwo.ca/cgi/viewcontent.cgi?article=9099&context=etd
*
```
.
```
#narcicism
2016 Oscars
— Mexican Best Director for The Renevant
— Mexican Cinematographer for The Renevant
2015 Oscars “So White” (87th Academy’s)
— Birdman was biggest winner
— Written, Produced, Directed, Cinematography— All Mexican!!
2014 Oscars (86th Academy’s)
— 12 Years a Slave won best picture
— Lupita won best supporting actress
— Gravity (Mexican cinematographer, director)
2013 Oscars (85th Academy’s)
— Jamie Foxx nominated for best actor
```
.
```
https://www.youtube.com/watch?v=ke_Syg4fj3c
Anger
Chris Rock
Dr. Jamal H. Bryant
Keep my wife’s name out your mouth
Romans 12:17-21
Vengeance is mine
```
.
```
#life
Life = Will to Power
Evasion = Numbing this Will
Methods = Ideology or Chemicals
x
Will (to do)
Desire (to be)
Senses 🐘 🦒 🦓 🦔 🐄 🐅
Only alcohol can keep man in hedonism
x
She should have died hereafter;
There would have been a time for such a word.--
To-morrow, and to-morrow, and to-morrow,
Creeps in this petty pace from day to day,
To the last syllable of recorded time;
And all our yesterdays have lighted fools
The way to dusty death. Out, out, brief candle!
Life's but a walking shadow; a poor player,
That struts and frets his hour upon the stage,
And then is heard no more: it is a tale
Told by an idiot, full of sound and fury,
Signifying nothing
Ambition — thou shalt be king
Meaning — signifying nothing
Pleasure — increases the desire?
No, alcohol merely inhibits mans will
Or numbs man whose will is too weak to climb
What is left are base instincts, sensualist, etc
x
0g.κοσμογονία,γ Mass/Energy
— Egypticism
— Roman
1f.ἡ ἔρις,κ Flux
— Frailty 🐑
— Robustness 🦅
2e.πρᾶξις,σ Catabolism
— Peace
— War
3e.ἄσκησις,μ Anabolism
— Décadence
— Culture
4d.∫δυσφορία.dt,ψ Plus Ça Change
— Memory
— Forgetfulness
5c.φάρμακον,δ HPA-Axis
— Opium
— Frenzy
6b.ομορφιά,β Exercise/Doping
— Pretentious
— Light footed 💃🏾 🕺🏾
7a.τάξη,α Performance 🎭
— Good & Evil
— Beyond Good & Evil
```
.
```
#shakespeare
Ambitious
— Macbeth
L’Homme Moyen
— Hamlet
Slacker
— Falstaff
```
Non plus ultra [^22]
[^22]: bards non plus ultra
.
```
#univariable
1.World views like “Jesus is the answer”
2.Virtually all ideologies
3.Punchlines, devices, etc.
Metaphysical comforters
```
.
Study lucky daye — Teija is a fan & she’d thus far been disappointed by Neo RnB
Of course she’s an old soul — yob 1995 — and her late 50s dad was a DJ
Tisha-like, but looks 10 years younger!!
.
```
#art
It’s art, and art only, that reveals us to ourselves
— Portrait of Mr. W.H.
To err is human
— Alexander Pope
All these things originate from that instinct which found in pain its most potent mnemonic. In a certain sense the whole of asceticism is to be ascribed to this
— What is the meaning of ascetic ideals?
```
.
```
#shultz
1.There should be an aristocracy
2.There should be mountains and valleys
3.Fresh brooks will always flow from high places to low
4.
5.
```
.
```
Psychology of money: the lesson here is..
— Anecdotes
— Man of science isn’t moved
— Successful
— Retired at 40
— Died at 92 with $8m
— Never trust retirement as achievement
— It’s frailty
— Patience as virtue
— Greed as Vice
Tremendous struggle reading this book
```
.
```
#adams
Adams was a dedicated diarist and regularly corresponded with many important contemporaries, including his wife and adviser Abigail Adams as well as his friend and rival Thomas Jefferson
```
.
```
Ambitious — Kim
L’Femme Moyen — Khloe
Slacker — Kourtney*
*PDA in S1E1 of both shows
```
.
```
80yo M
Presented: x 5 years (Insidious onset)?
— Palpitations
— Weakness
— Distance to bathroom
Recent:
— Otherwise a very active person
— Above issues lead to Dx: NHL/CLL.. 2 cycles of CHOP including last one in march
— Received several blood transfusions (Hb was 4 earlier in the year)
— Bone marrow biopsy: CD20 immunofluorescence pattern on H&E
— Received x? Rounds of chemotherapy so far
— Chief issue now is??
Surgery: 10/2021
— Intestinal Obstruction
— Hernia
Labs
— HGB 8.2 g/dL (Origin of presenting symptoms)
*
Symptoms: nodes, fever, night sweats, weight loss, tiredness
4 types of NHL (this case is CLL)
60 sub-types
Rx: slow vs. fast-growing, local vs. spread
Chemotherapy increases risk of other cancers (e.g. R-CHOP: well tolerated)
Affects 2% of Americans at some point in life, 4% of all cancers
Mostly between 65-75yo
5y survival is 71%
Staging CT-scan after 3 cycles for response
Of increased prevalence over last 29y
Window of opportunity: had surgery in 10/2021.
Did he have anemia then?
Received a blood transfusion before surgery..
*
Hb at surgery
```
.
Peter Thiel and [education reform](https://www.youtube.com/watch?v=WOEsVjqoOfA)
.
```
Mighty
— Imposing
— Lacrimosa
— What makes man, Mr. Lebowski?
Q&A
— Formulaic
— Lady, I got buddies who died facedown in the muck so that you and I could enjoy this restaurant!
Pleasing
— Circle of Fifths
— White Russian
— a “J” 🚬
— Bowling
```
#### 05/2022
```
#runnethover
1.First authors with Dorry as senior
2.My Stata class courtesy of Allan
3.What may I leave others?
A-List (Change), Company
— Musculoskeletal
— Drop outta school
— Found another institution
— Best your school in market cap
B-List (Being), School
— Cognitive
— Have been a sponge for nearly half a century
— Q&A: MD MPH PhD
— Killing it off!
— That we never ask questions that aren’t questions
— And instead proceed with hike to the mountain top :)
C-List (Abide), Idiosyncrasy
— Sensory
— Cassio Stereo sampling CDP 100
— 9 mile running at a pace of 7’-8’/mile
— Delights of the culinary kind
— Belgian beer
— Aged cheeses and snacking
— Chess: Kevin
1.Pretty clear what I’ve neglected
2.My natural home judging by Rugby, Academic, Musical glory
3.But then I made a Knights move :)
```
.
```
Santiago Alvarez Arango (This is my Guy!!)
*Free-text data using natural language processing (MGH.. informatics person, had developed medical extraction from the tests)
— Why Massachussetts
— Not enough EMR at Hopkins
— Or no persons with analytic skills for free-test data..
— Allergy specialist: drug hypersensitivity
— EMR records: evidence of hypersensitivity
— Immediate Vancomoycine Hypersensitivity reactions
1.Phenotype (HEIGHT)
— Different vancomycin phenotypes
— Focus on vancomycin infusion reactions (VIR), non-IgE
— New receptor Mas-related G protein (MRGPRX2) in cutaneous Mast cells
2.Exposure (SLOPE)
— Vancomycin is one of the most prescribed antibiotics (empirical, MRSA)
— Skin testing not validated
— Antihistamine slow inclusion may be preventive
3.Idiosyncrasy (INTERCEPT)
—
—
—
1.Describe EPI from EMR
2.Define skin protocol to distinguish non-IgE immediate to VIR (lower skin test threshold vs. drug-tolerant controls)
3a.Determine immunogenicity to vancomycin in recipients
— fully remotely
— Remote consenting (change in protocol), at home collection, amazon gift cards
— data on vancomycin history
— MRGPRX2 mechanism favored by low immunogenicity
3b.Assess. basophil responses in functional basophil assays in vancomycin tolerant subjects
— Functional
1.twenty minutes of interactive discussion
2.comments about the work
3.methodological issue
4.ask presenter questions
5.then maybe open for general discussion
VIR — moving beyond “red man syndrome”
Santiago et al came up with this term racially neutral term?
```
followup [^23]
[^23]: another potential r03 inspiration
.
```
#ulysses
2022/05/10
— Finally able to smile 😊 //comprehension
— Stately, plump, Buck… //anthropometry
— God, these bloody English //resentment
— Snotgreen, a new colour for… //neologism
— The mockery of it.. second-leg //hand
— Rage of Caliban.. //compadre
—
```
.
Canelé pastry cake
La Bohem cake 🧁
.
```
#innovation
Daniel Hayes
Met at Food Market
Wife left him during his rehab
Had head-on collision while driving
She’s not a gold digger, he insists
https://www.consensusortho.com/about/
Invited me to send him ideas
That I could have patents to my names
Although his focus is orthopedics
Kidney Transplantation wouldn’t be a bad idea
```
.
```
#taxes
Garnished salary
1.04/29 xxx
2.05/13 xxx
3.
4.
5.
```
.
```
#outliers
Malcom Gladwell
Classifies intelligence:
General and practical
Declares them orthogonal
Fails to see the interaction
Analytic intelligence causes
Independence early in life &
Yet dependence fosters relationships,
Skills one may call interpersonal,
Practical intelligence!
By what lucky strokes does one have both?
Teamwork with a perfect foil
Realism from day to day
Never asking questions that aren’t questions
Lack of opposition to moral problems of others
```
.
```
#accomplishments
1.Identified a consilient philosopher
2.Not the respected E. O. Wilson!
3.Have built a metaphysical system
4.Only to illustrate whence metaphysics comes
5.And what its destiny is: τάξη, same as realism
6.Resolved to return to the species essence:
7.Physical activity first and foremost
8.Adaptation is second-rate, reactive
9.Reminiscent of resentment
*
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ
5c.φάρμακον,δ
6b.ομορφιά,β
7a.τάξη,α
```
.
```
#kids
3-11 piano
12-18 track, field & pool. Buy/Rent Stadium
19-22 software, hardware eng, pre-med
23-27 medical school
28-32 orthopedic surgery
33-35 sports medicine. Use Stadium as Clinic
36-90 rehab medicine. Ditto those with injuries
Innovation
Patenting
Registering, inc.
```
.
Breaststroke
https://www.youtube.com/watch?v=rVaXro6fcEQ
.
```
#achilles
https://hbswk.hbs.edu/item/for-entrepreneurs-blown-deadlines-can-crush-big-ideas
Meeting timelines!
```
.
```
#howtoengage
The student, employee, colleague
Who have mastered the basics
And are very bored with
Stuff others are doing:
Create extra credit, leadership, etc
```
no followup thru & thus importancec of journaling! [^24]
[^24]: how to engage folks
.
```
05/24/2022
When I reach 200% of my move goal for the hundredth time (presently at 92), then I’ll refine my activity goals as follows:
1.Burning 1000 active calories
2.After accounting for a 400-500 baseline
3.Therefore I estimate the goal to 1500 calories
Let’s make this work!!
```
.
```
#butterfly
05/29/22
Maggie, a 48yo WF, says I’m a source of inspiration to her and her two daughters
She told the daughters about a man who decided to challenge himself and learn butterfly
x
Valentine (Redhead, Off White)
Natalie (Black girl)
Shared lanes with these two badass young ladies
At the end the black girl said I’m an inspiration
To her and to many others I’m not even aware of
Because I’m a black man who seems to be a
Professional and badass at butterfly
x
THANK YOU!
x
Ambition: Learn butterfly at 41yo
Meaning: Inspire others by my success
Pleasure: Badass body that could get them xxx
```
.
```
#iglesia
This music is indistinguishable
From kifunda church music
In Bwaise or Kawempe
How did we lose the samba?
Or the salsa, mariachi???
```
.
```
#sleep
My sleep position changed
When I started doing swim drills
Specifically the backstroke dolphin
I now comfortably sleep on my back!!
Preferably with dangling legs
```
.
```
#queenbed
L 6’1” (I’m two inches short)
W 5’0” (Blade of scapular)
D 6’6” (Half-foot short)
Explains why I snugly fit
Backstroke Dolphin position
Knees slightly flexed
Since legs are dangling
And my arms streamline
```
.
5 x 50s of each stroke = 100 laps = 1000 cals
.
```
#taxes
Matt 22:17
17 Tell us therefore, What thinkest thou? Is it lawful to give tribute unto Caesar, or not?
```
.
#### 06/2022
```
#jonbaptiste
The ultimate aesthetic statement
— New Orleans
— Julliard School
— Etc.
```
.
```
#mustdothis
Smithonian national museum o American history
Paiting o indians hunting bison
Bison
Indians
Mural
Painting
DC
```
.
```
#firstever10daystreak
05/25/22 — 06/03/22
🏊🏾♂️
Of single 1000 calorie workouts
Based on 13-18 swim sets
All strokes with back dolphin
Supportive diet of almonds, cheese, dates
Weight stable but with muscle growth
Loss of fat & appearance of contours & abs
```
.
```
#siblings
Sarah Babirye 01/12/63
Charles Nabongo 10/26/65
Andrew Kairu 01/03/67
Sarah Namukasa 04/19/68
Harriet Timulungi XX/XX/71
Patrick Muinda 06/07/74
Martin Muwaga 01/20/76
Abimereki Dhatemwa 04/17/80
```
.
Yonnie Busingye
Keith Kibirango
.
```
#busboys
Teija — a lot like Tisha (look alike & vibes)
Alana — other young chic, more energy, not a morning person kinda girl, standing in for Teija one specific morning :)
```
.
```
#breakthrough
May 18-26
— Four strokes
— Back Dolphin
— Increased burn
— Equivalent to 9MI run
```
https://www.youtube.com/watch?v=VfJ105NN68U
.
```
Heart rate
— 6 years worth of records
— Max 207 (01/07/2018 after 1592/1000 calorie swim)
— Min 30 (01/18 & 24/2018 after 1683 & 1350/1000 calorie elliptical)
— Started record-keeping in 11/2017 and this may reflect youth
— Both youth and unconscious energy produced these extremes
— The Max 207 is a result of energy
— And the Min 30 is a result of the Max 207 and cardio endurance/fitness
— VO2Max > 45 from 12/2017-01/2018
```
.
Who needs a dynamometer when
You have the Hopak?
Shouldn’t a doctors first question be:
The last time you performed the Hopak? [^25]
[^25]: hopak > dynamometer
.
Ambitious, Inherent&Driven
Rules&Guides, Mortal&Unsure
Slacker, Backstory&Numbed
.
```
#prankishness
1.Overture K. 620. Unlike other fugues
2.Wonderer 159. Respect of convention
3.Born to etiquette. Yet dancing in fetters
4.No revolutionary “shackles off my feet…”
5.Classical or at least a renaissance style
6.Illegal acts not aesthetically pleasing
7.Perhaps immoral acts also fit this bill
8.Therefore aesthetics is about social class
9.And this leads us to a damning realization
#alone4life
#nowalkoneggshells
#thishasendedmanyrelationships
```
.
```
#usurpation
Hamlet and Macbeth both portray usurpers as ruling kings, and in each case it is made clear that such rule is not to be tolerated either by the ruled or by the universe. Indeed, the fate of kings is tied to the order of the universe, and dissension and tension in one is reflected in the other. Yet, the issue is complex, and while the usurper has no right to rule, it is not always clear who has the right to prevent him from ruling. In Macbeth, the fate of the usurper is ordained, and his downfall at the hands of the wronged Macduff, who is the rightful king. Macduff has the right to retake his throne by force. The hesitation of Hamlet, much commented on by critics, may derive from the fact that while he knows Claudio to be a regicide, to kill Claudio would make him a regicide as well. In both plays, the usurpation of the throne leads to a more dangerous and uncertain environment in the land, and nature concurs by expressing through storms and other travails visited upon the people that a great political wrong is also a great moral and religious wrong
Reference:
https://www.lotsofessays.com/viewpaper/1691621.html
```
.
```
#usurpation
Antiquity: Greek, Viking
Romans
Christians
Philosophers
Schopenhauer
Wagner
Nietzsche
Freud
Tidier than neologisms by usurpers
Theomachy
Superior
Order of rank
Parricide
Regicide
Anxiety of influence
```
.
```
#swim
#sets
06/18/227
Set:
100yd breast
100yd butterfly
100yd freestyle
100yd backstroke
x6 sets
nose plug for backstroke
2:00:48
2400yd
583 calories
pace 5’02”/37 strokes/100yd
#goal
pace 2’00”/30 strokes/100yd
```
.
```
#divine
#meaning
#beast
#beast
But beauty, real beauty, ends where an intellectual expression begins
He is some brainless beautiful creature who should always be here in the winter when we have no flowers to look at
#meaning
Look at the successful men in any of the learned professions
#divine
Except, of course, in the Church
```
.
```
#swim
06/21/22
— set: 400y individual medley
— order: bbbf
— x4 on 06/20/22, x5 on 06/21/22
— consider x6 on 06/22/22
— develop habits around the medley set
— reduce strain on given muscle group
#imset
https://www.youtube.com/watch?app=desktop&v=p54US-IGqNo
https://www.swimmingcam.com/camera-systems.php#prod5
```
.
```
#symptoms
#prankishness
#comedy
#marktwain
#jonstewart
#bellweather
Its not the fresh prince that’s a threat to comedy
Its the crown prince!
Its the fragility of leaders!
Comedy doesn’t change the world
But its a bellwether
When a society is under threat, comedians are the ones who get sent away first
```
.
```
#prankishness
Nothing succeeds if prankishness had no part in it — Friedrich Nietzsche
Moderation is a fatal thing. Nothing succeeds like excess — Oscar Wilde
```
.
```
#homervshesiod
#kadivsgigi
#poetryvscatalogue
Have you ever read the Theogony? It’s a fairly short book, although a fascinating account of the Greek gods. The Iliad and The Odyssey are epic works. So that’s for starters. I would argue that the Iliad and the Odyssey are also just more interesting, being histories that had it all: war, murder, rape, betrayal, adultery, gods, witches, magic, victory, defeat, misfortune, revenge, vindication—you name it, it’s in the Iliad or the Odyssey. Each of them could make a fine mini-series!
— Bryan Roth
Because it’s rubbish.
I’m joking, of course. To an extent. But the Theogony isn’t as famous as the Homeric epics for a number of reasons!
Firstly, it isn’t a coherent narrative in the same way as Homer’s works are. It’s really more of an epic catalogue (which, as we’ll know from the catalogue of ships in Iliad 2, are everyone’s favourite parts of an ancient text… not).
It’s also significant that the Homeric epics document a very popular set of stories — the Epic Cycle was epic indeed! Battles at Troy? Enduring heroes struggling to get home? Mythical monsters? Everyone wants to hear about those! Can we really pretend that a blunt list of the 50+ children of ‘Nereus and rich-haired Doris’ is as gripping as Achilles telling Hektor he’d like to hack him up and eat his flesh raw?
It hasn’t been studied in the school system (ancient to modern) like the Homeric epics have been; that’s one of the main reasons for the tenacity of Homer’s works — they’ve been very popular for a long time! The Theogony just hasn’t been treated with quite so much affection, over the years… it isn’t as easy to read, nor necessarily as much of a pleasure.
The Theogony also just isn’t quite as impressive as the Homeric epics. It’s only about 1000 lines, compared to the Odyssey’s ~12,000, or the Iliad’s ~15,000.
— Amy Dakin
```
hilarious [^26]
[^26]: spontaneous debate 'twixt elementary school student & grad school professor
.
```
#agon
#gesture
#usurpation
#threatening
#smellstench
#listeningtodarkchild
— Wrapped up, tied up, tangled up
— Dawkins & Dawkins, 1998
— That beat provokes the gesture
— Harp arpeggios recall The Boy is Mine, 1998
```
.
```
#jerktaco
1.Jerk chicken burrito
2.Large oxtail
3.Meat patty
```
.
```
#gospel
#marcushodge
— Will Smith — Will, Chapter Fourteen: Boom, 10:04/-47:03
— That’s what caught my attention
— Its the brilliance of Marcus Hodge
— Now to investigate him a little
https://www.youtube.com/watch?v=noaURkEc6gc
```
.
```
#swim
#flip
https://www.youtube.com/watch?v=Nrn-vHXCoM8
```
#### 07/2022
```
Hard work beats talent any day!
```
.
```
#equation
#genius
#ancestry
#respect
#nopedigree
Nietzsche=Mozart
Freud=Beethoven
Claims of originality (Freud)
Or influence of descendants (Beethoven)
It is Beethoven’s descendants who lose interest in everything preceding him
Whether to blame this on Beethoven himself is irrelevant
He held Mozart in high esteem:
Cadenza for K. 466 Allegro
Blame his offspring?
```
something here [^27]
[^27]: originality
.
```
#mulenga
#dossier
#kingmaker
#kingceasor
#consulofvietnam
#minerals
#telecom #gemtel #itel
#business
#education
#tourism
#microfinance
#bafumbira #king
#kisoro
#kigezi
#ankore
#philanthropy
#banyakigezi #diaspora
https://kcu.ac.ug
Ceasor Augustus Mulenga
Charity Basaza Mulenga
Tress Bucyanayandi
04/09/2020
UGX 100M towards COVID-19 response plan
— Hands over a dummy cheque to office of prime minister Ruhakana Rugunda
— Tayebwa provides support as he holds gigantic dummy cheque
— All draped in yellow ties
“Throughout President Museveni’s campaigns in Kigezi and Ankole, the slender and shy-looking Mulenga has played a prominent role. On the eve of Museveni’s visit to Kisoro, he organized a fete in the municipality where locals ate and drank till morning”
#beastnomore
#surehethatmadeus
#withsuchlargediscourse
07/06/2022
1.It’s summer vacation (for many)
2.And beginning of the year (for us in medicine: day 1 is July 1st)
3.Makes impromptu meetings near impossible
4.So my goals is to walk you around to appreciate infrastructure (key for medicine!!!)
5.Visit again but with schedule to meet and agenda (calendars booked till June 2023)
6.Between September 7 — June 30 is best
— University: Rules of aesthetics
— Accreditation in the US
— Students then may visit for electives
— In return American universities may collaborate with $$
— Growth and much more
— Meeting with administrators tricky without these!!#
— Tripartite mission of education, service, research
— Shuttle service across campuses embodying tripartite mission!!!!!!!!!!!!!!!!!!!!!!!
— Virtually impossible to claim these without a sort of footprint
— Medical School, Teaching Hospitals, Peer-reviewed Publications
07/07/2022
— Employed Ariho & Charlotte before they got married
— Did these two meet in South Sudan??
— So this gives a firm background of at least 2 decades
#equation recruit individuals from old establishment
#genius claim them as founding fathers
#ancestry start a mythology backed with grand painting
#respect adhere to existing formulas for tripartite mission of university
#nopedigree they shall be known by their fruits: Matt 7:15-20 (also Matt 7 is simply awesome)
```
.
What is the low hanging fruit in America that Caesar can benefit from ?
Anything small to transfer to our country
.
```
#mulenga
#kingdom
#officialsong
Gramps Morgan
People Like You
The kingdoms Anthem
Caesar is Kuku’s about it
Listens to it every morning
It’s better you connect to Bluetooth
```
.
```
#29thfloor
Four seasons
— Caesar
— Favor
— Limits
— Gods plan
Those who exceeding the limits:
Moses
David
Caesar
Napoleon
Etc.
```
.
```
Learning communities institute
Virtual conference in October
Students, faculty, and stuff
Collaborate together and share
www.lci.org
Join as individual -$150, $600 for entire institution
Email Susan pulley for application fees
```
.
```
#ariho
Gordon
Charlotte
Curtis
Gabe
Emma
```
.
```
#swim
#fly 200m
Kristof Milak
WR: 1:50:24
```
.
```
#loseit
#lose.it
07/10/22
Milestone
— Exactly 6wk without calorie count:
— 05/30/22 was 365th day of use
— Weighed in at 178lbs on the day
— Qualitative documentation of all intake in the interim
— Comprehensive graphical summary available
— Photos may be found on 07/10/22 meals
— Now 1wk left to complete hiatus:
— Resume daily logging thereafter
— Strict adherence to calorie budget
— Holding debt not permitted ever again
— See if weight is still 178lbs on resumption!
— Does this mean my budget is too restrictive if still 178lbs?
Coherent
— Food 05/30/22 - 07/15/22: 700 calories over 45 days = 15.6 calories/day
— But this was scaled to 0.01 to allow me to “catchup” and return to schedule
— So its really 1,560 calories/day and a total of 70,000 calories over 45 days
— May reappraise this to 2,000 calories/day and 60,000 calories per month
— Empirical verification: 30,000 calories total on Activity App at each months end
— Nice, rounded, muscle building, familiar 1000/day burn!
Reappraisal
— Lets see how this is working on 07/15/22
— Then figure out how to consistently burn 1000 calories per day
— Swim, rower, other stuff, spare hip as much as possible
#justdoit
#justdo.it
```
.
```
#clinical
#guideline
#policy
#business
#academic
#service
#research
Great.
Make us appointments and negotiate a simple mou with any of medical departments or even yours. Your department could partner with KCU to pioneer organ transplant in Ug.
With Thomas support we can forge way forward.
With that we will Always be in Baltimore.
```
.
```
#uganda
#bill #act #transplantation
#jhu #facultypolicies #bluebook #goldbook #silverbook
#incentives
— Can any of these books offer me an incentive to push this agenda?
— Legal, guidelines, practice, research, collaboration
— KCU & JHU like MUJHU or recent?
```
.
```
#greeks
#transplantation
#uganda
#tayebwa
#bill #goal #act
#fuzzy: #ethics vs. #functionality ?
#primary clinical #secondary legal
#enable one market: regulated transplantation
#impede other market: organ trafficking, coercion of donors, etc
#aggressiveness #center
0g.κοσμογονία,γ Organ Transplantation
1f.ἡ ἔρις,κ Society vs. Patient
2e.πρᾶξις,σ Living Donation
3e.ἄσκησις,μ Deceased Donation
4d.∫δυσφορία.dt,ψ Waitlist Criteria
5c.φάρμακον,δ Immunological Barrier
6b.ομορφιά,β Other Barriers
7a.τάξη,α Performance Metrics
#gamma
#kappa
#sigma #networks
#mu #referral
#phi #conflicting
#delta
#beta
#alpha
Registry
— Logistics/intended: procure, HLA-match, transport, implant, survival, performance, reappraise
— Trafficking/unintended: information, asymmetry, market, price, trust, growth, conferences
— Model/template: regulation, databases, EHR, publications, ATC, other fields of medicine
“Hospitals across the United States are throwing away less-than-perfect organs and denying the sickest people lifesaving transplants out of fear that poor surgical outcomes will result in a federal crackdown”
— 1954, Boston. First successful solid organ (kidney) transplant in history: living kidney donor
— 1963, Pittsburg. First successful live transplant in history: deceased donor
— 1967, Capetown. First successful heat transplant in history: deceased donor
— rollout across N. America, S. Africa, Europe, Asia
— later rollout to Egypt, Nigeria, Kenya
— Uganda yet to join global transplant community
— Meanwhile patients with indications for transplantation have two options
— Tremendous resources for transplant tourism vs. death sentence
— Propose bill to remove necessity of transplant tourism and foster local access for all
— Improve longevity and quality of life of those who would otherwise be given a death sentence
— Model after S. Africa, culturally the closest success-story with locally relevant experiences
— Any existing laws on blood transfusion? How do these change with Part I: 3. Application?
Commentary
*MEMORANDUM
1.POLICY AND PRINCIPLES
— Motivation? Vague! Leaning towards ethics vs. functionality
— How about mortality? Death sentence for those without resources to travel?
— No Ugandan laws in place but templates available and must be stated at the outset
— Allows us to go straight to the nuances: choose NA, Europe, or Asia as template
— Explicitly declare what template we’ll go after and then proceed from there
Principle XX of UG constitution:
“State shall take all practical measures to ensure the provision of basic medical services to the population” Omit the “protect Ugandans from being victims of organ, cell and tissue trafficking.”
Lets be more functional than moralist, more alive than dull, more empirical than theoretical
2.DEFECTS IN THE EXISTING LAW
— Language inappropriate
— Can’t have defect where no law exists
— Prevention of trafficking can’t be motivation!
— Reducing transplant tourism to India can’t be motivation
— Microcosm of entire health sector: witnessing symptoms of problems
— Allow de-centralized innovation and let government stick to regulation of sector!
3.REMEDIES PROPOSED TO DEAL WITH DEFECTS
(B) designate Mulago National Referral Hospital as the pioneering transplant center
— Naive since a network of centers is key to success from onset (2)
— Should have member hospital centers as basis of Uganda Organ and Transplant Council
— With other stakeholders including GoU and maybe neighboring countries like Sudan, Congo, Rwanda?
— Local organ procuring organizations that may compete & collaborate (3)
— HLA typing, overcoming immunological barriers via swaps (4, 5, 6)
— Post-transplant care is lifetime and should not be local: not Kampala! (7)
— Expertise built at center-level by sending trainees to Univ. of Capetown (0, 1)
(G) establish standards for storage of harvested organs, tissues and cells
— This may be of very little relevance to solid organ transplant
— Here logistical barriers are the key concern: transplant within 8hrs of harvest!!!!
*PART I. Preliminary: removal, storage & transplantation vs. harvest, transport & implant
*PART II. Minister and transplant council vs. opo, logistics & surgeons
*PART III. Finances of council vs. steakholders: GoU, business & centers
*PART IV. Designation of transplant centers vs. all hospitals members, some voting members
*PART V. Additional requirements vs. from non-voting to voting member
*PART VI. Quality assurance: performance metrics, contract academic/statistical SRTR
*Part VII. Storage of organs, tissues, cells: transport not storage
*Part VIII. Transplant of organs, tissues, cells: ?????
*Part IX. Post-mortem of BDD: EEG? Post-mortem unnecessary cost
*Part X. Database and reporting requirements (Contract????): see Part VI — peer-reviewed
*Part XI. Offenses and penalties: legal stuff that is not my area of speciality
*Part XII. Miscellaneous: detailed study of South African laws
PART I.
— Provide for a system to ensure equitable access to transplantation services for patients
— This should be #1 rather than buried in text as if it were a “by the way..”
— Once logistics for this are set up then we can organically leverage the infrastructure to…
— Ensure traceability of organs, tissues and cells and recall procedures
— Reduce verbosity of document to these two elements (i.e., primary vs. secondary)
PART II.
— The Minister shall provide information and increase awareness about organ transplantation
— Opt in at time of national ID registration? That’s how America does it
— Family reserves power to override patients decision after death
PART IV.
28.Designation of transplant centers
— Mulago is designated as the pioneer transplant center
— This is a false start and should strongly be discouraged
— Because Transplantation at its core is all about logistics
— With resources that can’t be concentrated in Kampala
— Such as deceased donor organ at a given time
— And the most compatible match for that deceased donor organ
— Only nationwide registries at OPO level would minimize asymmetries of info
— Reminder: post-transplant care must be de-centralized to ensure lifetime access
— Can’t see how designating a pioneer center helps this cause
— Stifles innovation and merely delays progress
29.Application for designation as transplant center
— All medical centers should be represented in the Council
— Only those doing transplants may be voting members
— Ensures transparency of process towards membership
— Criteria for membership should be open, automatic and not for Minister to interpret
30.Qualification for designation as transplant center
— Two adjacent theaters; one for the donor and another for the recipient. Only necessary in the case of living donor transplantation, and only if the donor is in the same hospital
— Deceased donor transplantation never has such needs as organ harvesting or procurement is performed at the center where the donor was declared brain dead. This site is almost certainly never the same as the recipient center, since there is a waitlist and also HLA-compatibility issues that often lead to the logistical necessity of transporting a procured organ from one center to another
— Living donor transplantation on the other hand is often performed in two adjacent theaters. As such, this item uncovers unstated assumptions about deceased donor vs. living donor transplantation across the entire bill. These should be explicitly stated at the outset!
31.Designation by Minister
— There shouldn’t be any role for Minister in designating a hospital a transplant centers
— Open and transparent criteria are sufficient
— As such a hospital should automatically be considered eligible once it meets these
— Center should then be invited to (or should itself)) submit the application
— It should expect to be granted status unless criteria insufficient
— Process should be open to both voting and non-voting members
— (6) A hospital seeking to be designated to undertake cadaveric transplantation shall be required to get additional approval. Cadaveric is an out-of-fashion term. We now say Deceased donor transplantation. Also, this statement once again tells us that the unstated assumption is that living donor transplantation is the default mode where no adjective such as deceased donor is used. I believe this makes the bill vague and poorly worded and should be revised in its entirely to make the distinction between living donor and deceased donor transplantation from the outset.
32.Existing centers
— Quality/Performance metrics may render a center sub-par
— This should be grounds for temporary suspension of license
— Pending reappraisal of issues that led to suspension
34.Approval of banks
— For solid organ transplantation there is no such thing as “banks”
— Organs remain viable only within an 8 hour window of procurement
— The case for tissues and cells is different and this should be made clear
37.Education services to the public
— (4) Every designated transplant center shall produce or have available literature and media items that provide education and awareness creation for donation of organs, tissues, and cells
— For any of the outlined issues to be empirically based (i.e., scientific), Transplant centers will have to derived from peer-reviewed research. This item is lacking from the bill
— I propose that the database that is established for regulatory purposes should not only be used to ensure traceability of organs and prevent trafficking, but also be put to use in registry-based research with annual reports published in a peer-reviewed journal such as Makerere’s African Journal of Health Sciences
— Non-designated transplant centers and academic institutions should have access to registry data for research purposes. These data may be de-identified to comply with patient privacy
Part VI.
40.Organ Donation and Transport Quality Control System
(e) a database for all potential recipients and donors. I propose the name Uganda Network for Organ Sharing — UNOS. This serves as a reminder that the designation of Mulago as the pioneer center is misguided and potentially harmful to innovation and growth of a truly de-centralized network of organ sharing.
41.Quality assurance programmes
Internal audit or risk management. A competitive contract may be issued to academic or statistical agencies, carrying the name SRTR — Scientific Registry of Transplant Recipients. This registry will obtain regulatory and administrative data from UNOS and use it to quantify clinical outcomes such as death while on the waitlist, time from waitlist to transplant, allograft failure after transplant, immunosuppressive therapy used after transplant, primary graft non-function, survival after transplant, etc.
The SRTR will produce annual peer-reviewed publications that summarize key metrics. However, the SRTR should also make its data available to third-parties, who may also use it for academic and peer-reviewed research.
Only such an arrangement will ensure transparency, high quality appraisal of clinical outcomes, and center-level performance compared with the national average or some such benchmark.
45.Preservation of organs, tissues and cells.
— This may only apply to tissues and cells, never to organs!
48.Review of donor medical information
— Prior to the distribution of any organ, tissue or cell for transplantation, the registered medical practiional of a bank…
— In the case of living donor transplantation, this is meaningless since the donated organ will be in an adjacent operating theater to the one of the recipient
— So this mostly applies to deceased donor transplantation. As such, the entire bill remains somewhat confused about this distinction, which is not made at the outset.
PART VIII.
52.National waiting list
— Every patient indicated for organ transplant should be registered on the deceased donor waiting list
— However some registrants may proceed to living donor transplantation
— Thereafter they’ll be removed from the waiting list
53.Fair and equitable system.
— (5) Access to organs, cells and tissues shall be provided without regard to recipient sex, age, religion, race, creed, color or financial standing
— What about nationality?
54.Transplantation activity.
— (1) The retrieval and preservation of human organs…
— Lets be consistent with terms used
— Internationally, the term used is “The procurement… of human organs”
— (5) When a person has been confirmed brain dead by the team in care, the Council shall immediately be notified to send an independent neurological team to confirm the death for purposes of donation.
— This, again, only applies to deceased donor transplantation
— And this activity is not practical. An EEG is the standard way to define brain death and that can be electronically documented for the team at the local hospital. The patients relatives can review the evidence even without training (the local neurological team may explain the technical details)
— Family reserve the right to refuse to offer their loved ones body organs for transplantation
60.Preservation for transplantation
— (1) Where part of the body of a brain-dead person in a hospital, nursing home or other institution is or may be suitable for use for translation, the hospital, nursing home or institution shall preserve the body and transfer it to a designated transplant center for harvesting in accordance with this Act
— This may be prohibitively expensive. Typically a team of surgeons is sent to harvest the organs at the center where brain-death is declared. Only the organ is transported to the recipient hospital. This is cheaper and more cost-effective
— It is also logistically more meaningful since several organs may be procured from one deceased donor and the recipient hospitals for the various organs are unlikely to be the same since a waitlist is defined organ by organ
61.Harvesting of organs, tissues and cells
— Lets use the term procurement of organs
— National ID registration may be linked to organ donor status as done in the US
62.Retrieval procedure
— Lets be consistent
— Drop “harvesting”, “retrieval” from the entire bill
— Replace with “deceased donor organ procurement” throughout the document!
— With living donation the term used is “living donation”
— Again, let the entire bill from the onset distinguish between these two modes of donation!
66.Recipient followup
— Absolutely key for performance metrics
— System should transfer followup medical records to the transplantation database
67.Donation and transplant of organs from living donors
— The distinction between living and deceased donors must be made at the outset
— Issues pertaining to these two modes are often very distinct
— Entire bill should reflect that
PART IX.
78.Requirements for carrying out post-mortem
— Unnecessary cost
— Other means of assessing organ quality (i.e., donor risk index)
PART X.
— (3) Subject to the Constitution and Access to Information Act, 2005, information contained on the database may be made available to the public in a manner prescribed by regulations
— This is absolutely critical for transparency as well as outcomes research and quality control by third-parties as well as by regulators
XII.
93.Counseling of donors and recipients
— This is living donors
— Not applicable to deceased donors
— Reminder to distinguish the two
```
.
```
#theeconomist
#finalsubscription 09/01/99 - 07/13/22
#outgrownit
— great material on British grammar & marvelous style guide
— book reviews led me to Steven Pinker’s The Sense of Style
— outstanding Thinking Person’s Guide to Writing in the 21st Century
— however, I’ve reached a philosophical conclusion that kills its value:
— ambition, q&a, pleasure: that’s as many types of human stories as exist
*
— but will smith’s audio autobiography was so good an advert for audio media
— will thus give the economist one last run but purely as an audio medium
— I fully expect all the stories to fall under the three categories outlined below
— must remain open to this philosophical formula being wrong
— no problem whatsoever though I don’t see that happening
#ambition puff’d
#questionsanswers mortal and unsure
#pleasure a beast, no more
```
.
```
#kanyandahi
#toro
#mwenda
#nyangoma
#nyakato
```
.
Methinks, mistress, you should have little reasonfor that: and yet, to say the truth, reason andlove keep little company together now-a-days; the more the pity that some honest neighbours will not make them friends. Nay, I can gleek upon occasion.
.
```
#calculus
#heart
#running #walking #sedentary
* Myactivity
Running 04/17/22 (around B’more)
— 78min
— 165BPM
— 12870 beats
— 1084CAL
— 12870/1084=12 beats/cal
Walking 07/14/22 (to and from JHU)
— 25min
— 107BPM
— 2675 beats
— 122CAL
— 2675/122 = 22 beats/cal
Sedentary 07/14/22 (recumbent or seated)
— 44min
— 79BPM
— 3476 beats
— 48CAL
— 3476/48 = 72 beats/cal
* Textbook
— 72 beats/min * 60min/hr * 24/day = 72*60*24 = 103,680 beats/day
* Rescale
Running all day
— Empirically = 103,680/12 = 8640 calories
Walking all day
— Empirically = 103,680/22 = 4712 calories
Sedentary all day
— Empirically = 103,680/72 = 1400 calories
Weights (My typical day in 2021):
1 HR running
4 HRS walking
19 HRS sedentary
Average, weighted
(8640*1 + 4712*4 + 1400*19)/24 = 2253 calories/day
#running
#walking
#sedentary
```
.
```
#sean
#combs
#puffy #diddy #love
— consistent track record
— made folks rich & famous
— all by 23yo
```
.
```
#food
#ethnic
#report
07/17/22
— jerk taco is my #1 goto for Jamaican
— very reliable quality, authenticity
— but horribly unreliable and inaccessible
— no longer partner with door dash, grub hub or anyone
— sporadically closed on days they should be open
— officially never open on Sunday and monday
— testament to their quality: I’ve hang on despite…
— would say something similar about savior west African food
— however, the oiliness of the food is intolerable
— perhaps my favorite west African restaurant yet
— more is the pity that I have to let them go
— just assess the quality of their food a day later when no hungry!
— likelihood that sheer hunger has thus far sustained my interest
— end this today
— food market remains my #1 restaurant overall
— professional chefs and best of western/european cuisine
— also best street tacos, better than any Mexican ones I’ve had!
```
.
```
#loseit
07/17/22 09:57 hrs
— completed 50d break from logging
— hiatus “qualitatively” logged
— interim info logged on 07/15/22
— logged as .01 of whatever quantity
— lower bound “quantity” thus x100
— roughly 75,700 calories
— 05/30/22 thru 07/17/22
— 7wk, 7d/w, 49 days, 1544 calories/day
— assess weight two days from now
— very distorted from juice & coffee
```
.
```
#grubhub vs.
#doordash u screwed
#brazil gourmet market
07/17/22
30% cheaper with amazon/grubhub vs. door dash
Pão de Queijo #1 $4.98 vs. $6.10
Pastel - 6 pieces #2 $10.90 vs. $13.20
Mixed appetizers $13.90 vs. $15.70
Brazil Gourmet Burger $9.98 vs. $12.10 *
Picanha sandwich $14.90 vs. $16.90
$91.03 (including $6.50 tip)
$71.41 (including $8.00 tip)
Picanha Samdwich
Pasteís cod fish
#juiciest burger ever*
#picanha try beef
#appetizers nice
```
.
```
#theeconomist
#cultural #references
#humor #dry
The Economist
07/14/22
The course of cultural regeneration never did run smooth
#specific
#hardcode
#generalize
```
.
```
#words
Concupiscence
Tumescence
Detumescence
```
.
```
#favorite
#soul puff’d
#mind unsure
#body surely..
From 07/05/21
1.Art glorifies human errors
2.Does the realism of The Wire qualify as art?
3.We are seduced by flawed characters, especially the West Baltimore cast:
4.Omar, Snoopy, Senator Clay Davis, but also by the Police Department…
5.So why, then, does season 5 fail? Why does McNutty seem contrived?
— It abandons reality
— Becomes intellectual
— Ultimately preachy
— Moral of story.. of the show becomes explicit
— Here we have the viewers intelligence greatly abused
Viewers can connects dots from the chaos of reality: 1-4?
Today 07/20/22
#decadent S5: from soul to mind
#contrived a symptom of above
#basis of all aesthetics
###
###
###
#atheism
Atheists are anything but aesthetes
They are moralists against religion
Better their reference than other
Vs.
Irreligiousness of Artists. — Homer is so much at home among his gods and is as a poet so good natured to them that he must have been profoundly irreligious. That which was brought to him by the popular faith — a mean, crude and partially repulsive superstition— he dealt with as freely as the Sculptor with his clay, therefore with the same freedom that Aeschylus and Aristophanes evinced and with which in later times the great artists of the renaissance, and also Shakespeare and Goethe, drew their pictures.
#aesthetics
```
.
```
#frailty
Our president is growing old. Instead of attending to day-to-day issues. He’s talking about the future. — Derreck Kabatsi
```
.
```
#nia
#grants
#r21 #r33
```
Transformative artificial intelligence and machine learning based strategies to identify determinants of exceptional health and life span (clinical trial not allowed)
Phased Innovation [Award](https://grants.nih.gov/grants/guide/rfa-files/RFA-AG-23-033.html?utm_source=NIA+Main&utm_campaign=1cc51c8d8d-FOAS_June2022&utm_medium=email&utm_term=0_ffe42fdac3-1cc51c8d8d-18515504) [^28]
[^28] journaling!
```
#aiko
— kodaira, RN
— jhenè, Musician
Flex
on my X
In my model X
Lil-bitty body, modelesque!
— H.E.R.
```
.
```
#exemplar
#colditz, graham
MBBS, 1979 MBChB, 2004
MPH 1982 MPH, 2007
DrPH 1986, PhD 2025
MD 1998
```
exemplar? [^29]
[^29]: exemplar?
.
The [choice](https://www.economist.com/britain/2022/07/21/the-choice-between-rishi-sunak-and-liz-truss) between Rishi Sunak and Liz Truss from The Economist
.
```
#truth
```
To this end was I born, and for this cause came.I into the world, that Is should bear witness unto the truth. Every one that is of the. Truth heareth my voice.
Pilate saith unto him, What is truth?
— John 18:37
.
```
#art
#commodity
#davechapelle
— Artists should never behave like a commodity
— Despite recent history wherein art has fetched millions/billions
— Those responsible for this inflation are philistines wishing to be considered cultured
Q: why would I pay so much for shakespeare, Mozart, or Nietzsche?
#nuance
#required
#nocensor
```
.
`#freedom`
Freedom means different things to different kinds of people. To the warrior it is frequent opportunities to test ones strength against a worthy adversary. To the average man it is the opportunity to fully express oneself — i.e., ones faith, ones hope, ones love — as one awaits the eternal comforts of their cherished ideal. To the slacker it means unlimited access to narcotics or anything that offers escape.
.
```
#race
#spring
#worldchampionship
#jamaica
1.Shelly-ann fraser Pryce (14y since her first global title, is 35yo)
2.Shericka Jackson*
3.Elaine-thomspon hera
*Triple threat to be watched
— Youngest & personal best
— Winning at 100m, 200m, 400m
— Unprecedented
```
#### 08/2022
```
#nietzsche
#quantum
#relativity
The physicists believe in a "true world" after their own kind; a fixed systematizing of atoms to perform necessary movements, and holding good equally of all creatures, so that, according to them, the "world of appearance" reduces itself to the side of general and generally-needed Being, which is accessible to every one according to his kind (accessible and also adjusted,—made "subjective"). But here they are in error. The atom which they postulate is arrived at by the logic of that perspective of consciousness; it is in itself therefore a subjective fiction. This picture of the world which they project is in no way essentially different from the subjective picture: the only difference is, that it is composed simply with more extended senses, but certainly with our senses.... And in the end, without knowing it, they left something out of the constellation: precisely the necessary perspective factor, by means of which every centre of power—and not man alone—constructs the rest of the world from its point of view—that is to say, measures it, feels it, and moulds it according to its degree of strength.... They forgot to reckon with this perspective-fixing power, in "true being,"—or, in school-terms, subject-being. They suppose that this was "evolved" and added;—but even the chemical investigator needs it: it is indeed specific Being, which determines action and reaction according to circumstances
Aphorism 636
Will to Power
```
.
All art glorifies or vilifies some value system
For this reason it exposed the artist
Tyler Perry is an open book
Shakespeare less so
Less so is it!!
.
08/07/22
An older woman (always an older woman) remarked that I swam like a Dolphin!
I approached her in the butterfly stroke, half-way on my 4000YD x10 set IM swim
Today I achieved my weight goal of 170lbs, set on May 30, 2021
.
#loseit
#colories
08/07/22
— 170 lbs goal achieved
— Budget automatically updated
— From 1710 CAL/day to 2459 CAL/day
— 1710 only catered to BMR (10*79+6.25*180-5*42+5)
— 749 is average calorie burn expected per day
#mifflin
#equation
.
```
07/09/19 at 08:25 HRS
— Subtle breakthrough
— Reality vs Metaphysics
— Later at 17:46 HRS
— Attribute it to Sapolsky
— But Sapolsky picked lNietzsche
— From 3 —> 5 —> 8 variables
— That’s the metaphysics, the error
09/18/19 at 18:41 HRS
— Inflection
— Move kappa*
— Over to beginning
01/28/20 at 09:00 HRS
— Naivety
— Ontology
— Epistemology
— Differentiation
— Ranking
03/12/20 at 16:23 HRS
— Raphael rooms
— Testing my framework
— Only worked with kappa to left*
```
.
```
#longfeet
#raphael
#metaphors
#poetry
#art
Raphael has long feet
Dancing spirit from
Antiquity to Christianity
Brings these into harmony!
Now this is princely freedom
```
.
```
#cuddle
#struggle
That’s it!
For me a
Cuddle is a
Struggle :(
```
.
#schoolofresentment
https://www.youtube.com/watch?v=1HdnjTCMzpg
The fact that I’m first hearing of Hazel Scott from Alicia Keys on 08/18/22 just might lead me to revise my stance on the school of resentment
.
`#breakthrough`
08/19/22
— monotheism
— ingredient
— tension
— narratives
— consequence
*
— theogony?
— alternative?
— necessary?
— symptomatic?
— overflowing?[^30]
[^30] dimentionality reduction
.
Monk’s Cafe:
— S. Broad St
— Market St
— 16th
.
Mark lives exactly 10 miles from me!
.
De Ranke Guldenberg 8%
https://www.monkscafe.com/bottles/
.
Idiom: The devil is in the details
Antithesis: Line of best fit (mean)
Synthesis: De devil is in de tail (spread)
.
```
*!lazy
#beauty
0g.κοσμογονία,γ theomachy morals
1f.ἡ ἔρις,κ dionysian beyond
2e.πρᾶξις,σ r2 olympics zarathustra
#ugly
3e.ἄσκησις,μ temples gay
4d.∫δυσφορία.dt,ψ #i. pessimism dawn
5c.φάρμακον,δ Ho frenzy human*
#aesthetics
6b.ομορφιά,β elderly untimely
7a.τάξη,α X=0 youth tragedy
*!faithful
#II 96 on 08/17/22
— protestant church: spread with startling rapidity
— old time creed: costly masses, symbolism, pomp & ceremony
— leisure? oremus nos, deus laboret!
```
.
```
*!evolution
#beauty Inference
0g.κοσμογονία,γ universe
1f.ἡ ἔρις,κ vibes
2e.πρᾶξις,σ r2 muscle
#ugly Lincom
3e.ἄσκησις,μ atrophy
4d.∫δυσφορία.dt,ψ #i. hpa-axis
5c.φάρμακον,δ Ho psilocybin
#aesthetics Coefficients
6b.ομορφιά,β gerontology
7a.τάξη,α X=0 youth
*!metaphysics
#adm I #2 on 08/18/22
— 0.cosmology,γ
— 1.ecology,κ
— 2.biology,σ r2
— 3.maths,μ error #1
— 4.philosophy,ψ #i. result #1 (de devil is in de tail)
— 5.tech/rx,δ Ho fix #1
— 6.art,β narrative #1
— 7.life,α X=0 offbeat #1
```
.
```
*!alicia
#beauty Inference
0g.κοσμογονία,γ universe
1f.ἡ ἔρις,κ vibes
2e.πρᾶξις,σ r2 muscle
#ugly Lincom
3e.ἄσκησις,μ atrophy
4d.∫δυσφορία.dt,ψ #i. hpa-axis
5c.φάρμακον,δ Ho opium
#aesthetics Coefficients
6b.ομορφιά,β gerontology
7a.τάξη,α X=0 youth
*!keys
#adm I #3 on 08/18/22
— chapters covering first 2 albums top-notch
— but later chapters are woke & dull (egypticism):
— music, lyrics, activism
*
When I saw Alicia at the MTV Video Music Awards, the only thing I noticed was her hair and outfit. She looked like that woke vegan auntie who travels to Africa every year. I’m into it! Her skin is fresh and beautiful. Why would she even need makeup?
*
— the “big” questions
— even answers!
— sisterhoods
*
Hazel Scott, Progression in C# Major
https://www.youtube.com/watch?v=LmujUkwU19I
https://www.youtube.com/watch?v=1HdnjTCMzpg
```
a neat reference [^3]
.
```
#noseplugs
#noseclips
#technique
#backstroke
08/23/22
Backstroke - Nose Plugs
#noseplugchamp
https://www.youtube.com/watch?app=desktop&v=qD4u2xwnt4E
Missy Franklin
https://en.m.wikipedia.org/wiki/Missy_Franklin
```
.
`#aiko`
3pm-8pm, 08/27/22
6901 Barrett Ind, Bethesda MD
.
`#loseit`
Mifflin formula (men)
5 - 5*AGE + 6.25*HEIGHT + 10*WEIGHT
Or
166*MALE - 161 - 5*AGE + 6.25*HEIGHT + 10*WEIGHT
.
One can only be silent and sit peacefully when one hath arrow and bow; otherwise one prateth and quarrelleth. Let your peace be a victory!
.
The happiness of man is, “I will.” The happiness of woman is, “He will.”
.
`#LDA`
Class only available 1st term
.
`#poolparty`
6901 Barette Lane
Off Bradley boulevard
Mere 5min from Wisconsin
Leffe blonde x 3
Westmalle dubbel x 2
Duvel x 2
Rocheford 10 x 2
Orval x 2
WoB & Silver x 4 pack cans (Tripel-style)
Hefeweizen x 2 (Weihenstephaner)
Dogfish IPA 60min x 2
.
```
#student
Calvin Mathew
South Florida
High school
Interested in mentorship
Organ transplantation
American Heritage Schools finalist for ISEF (1).
(1) https://www.miamilivingmagazine.com/post/american-heritage-schools-claims-top-honors-in-stem-competitions
```
.
`#perfection`
Raquel Roxanne "Rocsi" Diaz
.
```
#universalconstants
08/29/22
Marx —> Class Struggles
Nietzsche —> Will-to-Power
Einstein —> Speed-of-Light
```
.
Paris Hilton
https://www.youtube.com/watch?v=wOg0TY1jG3w
.
```
#turkey
08/29/22
googled: Turkish renaissance
motive: quality of medical services, aviation, film
1.Live donor liver transplantation: Johns Hopkins sent Nabil Dagher to Turkey for a year for an apprenticeship
2.Turkish airlines: spokesmen and women are americas A-list celebrities including…
3.Another self: unremarkable Turkish limited series on Netflix. The quality of the cinematography and editing is world-class. But its really the dialogue that has moments that say something profound about the people, the nation.
###
Turkey has witnessed sustained economic growth during the last ten years, with the country achieving average growth of 5% during 2002-2012, despite the fact that it is surrounded by neighbours facing political or economic crisis, representing a rather unstable external environment. Among its neighbours, Cyprus and Greece were deeply affected by the financial crisis that hit the Euro zone while Syria is being ravaged by civil war, and Iraq is unstable. The latest demonstrations against the Turkish government’s plan to redevelop Gezi Park turned into nationwide protests, particularly by the youth, and constitute a warning message that the views of the youth of Turkey require serious consideration. An important fact that cannot be overlooked is that the youth movements in the region were the main antecedents for substantial revolutionary changes.
Despite the need for caution in assessing Turkey’s economic prospects for the coming years, the country’s success in achieving strong growth so far, amid regional instability, is worth contemplating. In fact, Turkey lacks natural resources such as oil and natural gas that neighboring countries have. However the country generated growth by focusing on enhancing industry, services and tourism, which helped boost its competitiveness. The tourism sector attracted more than 36 million tourists in 2012, rendering Turkey one of the top tourist destinations worldwide. However, this level of growth requires more than reliance on market forces and official development assistance. In order to obtain long-term benefits for growth and poverty alleviation, Turkey engaged in private sector development and structural reforms that enhanced its education and healthcare services, placed stricter controls on public finances and made progress towards combating corruption. Through effective support policies that promote financial resilience, investment, productivity growth and technological change, the country has integrated itself into the global economy and has become one of the major foreign direct investment (FDI) recipients in the Middle East region. (It was rated as the 13th most attractive FDI destination in the world in A.T. Kearney’s FDI Confidence Index 2012).
At the macro-level there is a modern infrastructure with highways and railways that link Ankara, the capital, with other parts of the country. To stimulate technological development, the country has been transformed into an important higher education hub that attracts an increasing number of university students from Asia and Africa. Effective university-industry collaboration has contributed to providing domestic industry with its needs in important fields, including aeronautical technology, IT and electronics. The country has invested heavily in technology development zones with 34 operational technoparks and 16 under construction to support research and development activities and attract investment into high technology fields. Turkey has focused on environmental technology aimed at generating sustainable energy that benefits from wind and other renewable energy resources, enabling it to compete globally in innovative green technologies and products. Furthermore, the country has succeeded in solid waste management, turning Ankara into a safer and greener place to live. Methanol gas and organic waste are used to generate electricity and to produce fertilizers for agricultural products, including strawberries, tomatoes and orchids.
Turkey’s trade and investment strategy is based upon a strategic ‘opening up’ to emerging markets in Asia and Africa and the Middle East, with low entry barriers. Turkey benefits from a Customs Union agreement with the EU, and free trade agreements with several countries including EFTA ( European Free Trade Association) member countries Switzerland, Norway, Iceland and Liechtenstein, and also Egypt, Jordan, Morocco, Palestine, Syria, and Tunisia. This has contributed to growth and increased productivity in both the industrial and services sectors, including green technology and IT.
These achievements would not have been possible without the strong political will of the policy-makers to attain a turnaround in Turkey’s economy and move forward with its EU accession process. They embarked on an incremental process of institutional building and reforms that started in 2003 and aimed at addressing Turkey’s structural problems. The reforms had a positive impact on the banking sector, the management and control of the public budget, and directing domestic investment towards infrastructure, health, education and technology. In the field of education, concerted efforts by the government and education institutions aim to bridge the gender gap and enhance the quality of education and higher education.
A consistent and incremental path of institutional reforms has led to robust economic growth, increased resilience to the global economic crisis and a remarkable surge in exports, reaching $153 billion by the end of 2012, up from $36 billion in 2002. Most notably, Turkey was registered on the world economic scale as the 16th largest economy in the world, and as 6th largest economy when compared to EU countries in 2012.
Turkey’s remarkable achievements show that facing the challenge of enhancing international competitiveness and achieving sustainable growth could be met by combining national support policies for investment, productivity growth, and technological change (based in high value-added sectors) with emerging opportunities in the global economy. That is not to say that Turkey has developed a policy for development that can be replicated everywhere. Any process of reform and private sector development MUST take into account the large differences amongst countries and their individual, unique characteristics. Nevertheless, general policies could be considered – as common features among countries do exist – especially where these can be translated into country-specific policies.
```
.
```
#survivingtranslation
08/20/22
1.Marx in Communist Manifesto
2.Nietzsche in Attempt at Self-Criticism
3.Of whom else may this be said?
```
.
```
*!use&abuse
#monumental
0g.κοσμογονία,γ history
1f.ἡ ἔρις,κ* class
2e.πρᾶξις,σ r2 freeman
#antiquarian
3e.ἄσκησις,μ slave
4d.∫δυσφορία.dt,ψ #.i uninterrupted
5c.φάρμακον,δ Ho revolution
#critical
6b.ομορφιά,β epochs
7a.τάξη,α X=0 ranks
*!history
#adm IV #5 on 08/20/22
— action & struggle
— conservatism & reverence
— suffering & deliverance
```
#### 09/2022
Bruno Bauer
Karl Marx (thesis advisor: Bruno Bauer)
Friedrich Nietzsche (admirer of Bruno Bauer)
.
May flights of angels sing thee to thy rest!
.
Life can be an utter nightmare, devoid of pleasures and amounting to nothing more than a rigorous course of turmoil for damned souls, so once in a while you need a reminder that there might be more to this realm, and this is one such reminder, in beer form. Looks like a snowglobe, filled with various films and floaters. Smells like warm grain and banana. Powdery feeling, dry, banana and clove and honey, sweet but w/ burps of fuel sitting at 8%. Thanks for the brief reprieve you Belgian wizards
.
Indeed, it was by keeping her official alter ego as vague as the unwritten British constitution, and her private persona hidden away altogether, that Elizabeth II became the most successful sovereign since Victoria, bringing relevance to a feudal institution that was 200 years past its sell-by date
.
Mirage: Dan Draper, Betty Draper, and their beautiful kids
I guess may viewers will idealize this Apollonian image of perfection
But the Dionysian details that emerge from Madmen refute this picture
Now, should we talk about the perfect balance between Apollonian and Dionysian?
Of course not! Its all silly metaphysics
Bottom line
1. Bring in an account (industry)
2. Winning a grant (academia)
3. ???
.
```
David Simon
Like a modern day Homer
Has too many war stories to share
Simply no room for backstories
Except what might be surmised
From dialogues strewn throughout
Breaking bad also has none of this
It’s Sopranos & Madmen
That suffer from this fault
With their Freudian formulae
On which all stands or falls
Freud is a metaphysician
And so anything built on
Psychoanalytic formulae
Can be expected to last
Very briefly, Annie Hall notwithstanding
```
.
```
Uncle, you always spoke to me like an adult. I liked it!
08/15/22
Kevin Kayongo
WoB, Bethesda, MD
```
.
Survival analysis
09/12/22
Hi all,
I have got a few questions since Friday, on how to calculate $P(\Delta = 1)$ in HW1, so I am sending out a hint. Use the following formula ($T$ and $C$ are random variables, $t$ and $c$ are dummy variables, $\int_{a}^{b}$ means integrating from $a$ to $b$, $f_T$ and $f_C$ are the pdfs):
$$
P(\Delta = 1) = P(T<= C) = \int_{0}^{ub} \int_{0}^c f_T(t) \times f_C(c) dt dc
$$.
Note that the outer (first) integral is with respect to c, and the inner integral is with respect to t.
I won't be able to cover this in class further, but I found this thread of discussion providing some details/insights:
https://math.stackexchange.com/questions/2915476/how-to-calculate-pxy-of-two-random-variables .
Best,
Daisy
https://math.stackexchange.com/questions/2915476/how-to-calculate-pxy-of-two-random-variables
.
Req 92179: Sr. Research Assistant is right out of a Master’s program with no additional stats or clinical experience.
Req 91861 or92179: Research Data Analyst if Masters and 1 year paid experience or a clinical background.
.
Favorite song? Teach my hands to war — Marvin Sapp/Aaron Lindsey
Favorite bible verse? Psalm 144
How to simulate bipolar:
09/13/22
1. Starve
2. Espresso
3. Beer
4. Music
5. Dance
.
The wire >>>> Mad men
.
```
Mad men
09/14/22
1.Wikipedia reduces it to “identity” (aka being)
— Don Drapers fake identity
— Selling products with fake identity
— Unfaithfulness and fake marital identity
— Being a woman, black, etc. and the identity that comes from social rank
— This is all metaphysical BS
2.I say as follows:
— Peggy Olson becomes
— Starts off as a fledgling, not-so-pretty, mere secretary, who has been shit upon
— But she is never resentful (i.e., is not a victim, but an willful agent like any of the men/bosses/masters)
— Only gets stronger, harder, faster, prettier, more powerful, more desirable, more impressive
3.Art rightfully replaces morality as the metaphysical activity of life
— In review #1 above morality is the undertone (i.e., the mores of the 1960s)
— Even the creator of the show thinks this way
— Yet in review #2 art, insofar as this show depicts the aesthetic phenomenon of becoming, is overtone
— Worth noting that the creator doesn’t see it this way
4.Flashbacks and backstories
— This attempt to explain being
— Why is this character like so and so?
— Oh, look to their childhood! Freud and such shit!!!
— And that’s why Don Draper is only interesting in the first two episodes of season 1
— He is a strange mysterious figure, ambitious, but alas already a genius at work
— Now, when the flashbacks and backstories unveil his being its all downhill from here
— Similar decadence in the quality of writing cross-talks to Peggy Olson
— Now we know where she comes from and what happened to her child
— Then there’s the priest who gets into her grill and starts to preach about sin and eternal damnation
5.How does Mad men pull it off?
— I guess its the honest writers
— They are honestly metaphysical and moral
— But also at times honestly awed by becoming
— And so its all in there, as it is in life!!!
```
.
Nyege Nyege and its origins are truly a rebirth out of the spirit of music!
.
```
Sapolsky
Congential: Adrenal*
Ecology: Hormones
Behavior: Neurotransmitters
*
Congenital adrenal hyperplasia (CAH)
Androgen insensitivity syndrome (AIS)
The entire spectrum of LGBTAI+
```
.
```
Vogue
Khaby Lame, the Fresh Prince of TikTok, Is Launching His First Collection With Boss
With almost 141million followers on TikTok and just over 78 million more on Instagram, Khaby Lame is one of the most followed social media stars in the world. And how has the 22-year-old Italian achieved this? Chiefly through silently satirizing the ridiculous content that other people post in order to become more followed on social media. Now that really is meta.
“The funniest part of my content is the silence,” said Lame over WhatsApp yesterday: “Silent comedy speaks a language that everybody in the world can understand.” By doing in the digital age what Charlie Chaplin, Harold Lloyd, and Buster Keaton once did during the dawn of cinema, Lame has gone from unemployed factory worker (he started posting after being laid off at the start of the pandemic) to a man who rubs shoulders with global icons like Leo Messi: “Meeting him is one thing I still can’t believe happened.”
From 6 pm tonight in San Babila, Milan, Lame will launch his first-ever capsule collection with Boss before throwing a party later at the city’s most famous nightspot, Plastic, so his locally based fans will have the chance to see him IRL. “There will be a giant Khaby,” said Lame. “We will take Polaroid pictures. I will arrive on a scooter flanked by a team of BMX riders wearing the collection. It’ll be amazing!” Doubtless many more will check in via their screens too. Lame says that around 15% of his audience is from Italy, 25 % is from South America, and 30% is from the US*.
The Boss x Khaby collection is a tight ensemble including varsity jacket and hoodie. One surprising inclusion—and the reason for that ‘giant Khaby’ promised at the reveal later—is a Khaby doll designed to be used as an accessory or an attachment that can be snapped onto the garments. Huh? Lame explained: “When I was a kid I had an action figure of Vegeta from Dragon Ball and I’ve been collecting action figures for years. I’m really passionate about action figures and anime characters. And now to see that there is a doll of myself and there will be an event to launch it along with the collection… I’m super excited.” Lame's capsule will be on sale exclusively in Boss’s San Babila store today before going on global release tomorrow.
As well as pumping out content with his team every day, and thinking through his next capsule—“I definitely want to do another one”—Lame is also focused on learning English and plotting his future moves. He said: “I want to be an actor, like Will Smith. He started with comedy, then he made more dramatic movies. He’s done all sorts of films, he’s a great actor and I would like to be like him. In fact my passion for acting started with him, watching him in The Fresh Prince of Bel-Air. Watching that show, I realized that I also wanted to make people laugh.” Lame is certainly doing that on social. And with that 200 million-plus audience already behind him it seems a no-brainer that we’ll see him on bigger screens soon enough.
*explains the unapologetic Brazilian jersey on an Italian: incroyable!
```
.
```
Influencers
09/15/22
Nietzsche —> Elite
Freud —> Modern
Marx —> Worker
```
.
```
Villain
09/15/22
1.Uber rider’s first words to me
2.You look like a villain out of a movie
3.Don’t at all fit in this environment
4.And look suspicious, dangerous
5.Not sure what it is but maybe:
— debonaire look
— the fancy shades
— very casual look
— shorts, t, flip flops
— je ne sais quoi
x Nice conversation ensured
x Told him girls say I look humble
x But later say my look is misleading
x Also told him I loooove food
x En route to Jerk Taco
1.Asked for my number
2.Former restauranteer
3.Nigerian by origin
4.Wants to invite me
5.Looking forward
Chukwuemeka 4.92
Abimereki 4.97
```
I see no reason why thou shouldst be so superfluous to demand the time of the day
.
Weight
09/18/22
200.00 lbs
.
Yassss[!](http://www.math.wm.edu/~leemis/chart/UDR/PDFs/Uniform.pdf)
.
```
#drinks
09/19/20
1.Have 43 servings
2.Should last till 11/01/22
3.Review this 50 days from now!
```
.
Many a time and oft, many a morning hath he there been seen
1. Julius Caesar Act I Scene I
2. Merchant of Venice Act I Scene 3
3. Romeo and Julie Act I Scene 1
.
The fault, dear Brutus, is not in our stars / But in ourselves, that we are underlings
.
The cause is in my will, I will not come!
.
For mine own part it was Greek to me
Men at some time are masters of their fates. But in ourselves, that we are underlings. But, for mine own part, it was Greek to me. Let's carve him as a dish fit for the gods.
Julius Caesar A1S2
.
```
#books
09/20/22
This is a revolution!
Prefer audio to reading
Be a selective principle and..
Only choose the very best recordings
```
.
There is no disgrace in choosing tradition over growth. Other countries seem to do just that in their revealed preferences. But Italian per capita income is easier to live on when there is also Italian weather. Japanese stagnation is not so bad when there are also Japanese crime rates. If the UK embarks on the economic trajectories of those countries, what is its cushion?
.
```
#Epilepsy #Metaphorical #Prosaic
09/20/22
1.Julius Caesar
2.Dostoevsky
Cassius
But, soft, I pray you; what, did Caesar swoon?
Casca
He fell down in the market-place, and foamed at
mouth, and was speechless.
Brutus
'Tis very like; he hath the falling sickness.
Cassius (metaphorical)
No, Caesar hath it not; but you and I,
And honest Casca, we have the falling sickness.
Casca (prosaic)
I know not what you mean by that, but I am sure
Caesar fell down. If the tag-rag people did not clap him
and hiss him, according as he pleased and displeased
them, as they use to do the players in the theatre, I am
no true man.
```
.
```
Belgian Ales
09/20/22
#Ale
1.Blonde: Trappiste Westvleteren 95% * Malt (Beginners) ~ Lager
2.Pale: Orval 93%; (Strong): Duvel 94% * Balance: Hops/Malt
3.IPA: Chouffe 93%, Duvel Triple Hop 93% * Hops Mostly
#Trappiste
4.Dubbel: Westmalle 93%
5.Trippel: Van De Garre 97% (+ Unibroue, Westmalle, Karmeliet, Curieux)
6.Quad: Rochefort 10 99% (Westvelteren 100%)
#Sour
7.Oude Gueuze: Tilquin 98%
8.Flanders Red: Rodenbach 99%
9.Lambic: Oude Lambik 3 Fonteinen 91%
```
.
To bear steadfastly the reverses of fortune
.
Andrew Lanham
https://hls.harvard.edu/faculty/andrew-lanham/
Six degrees!
https://themarginaliareview.com/shakespeare-contra-nietzsche-andrew-lanham/
How he got my attention:
Philosophers and literary critics alike, from Walter Kaufmann to Jacques Derrida to Harold Bloom, routinely treat Shakespeare and Nietzsche as ciphers for one another, as if they were a strange form of conjoined twins, their tragic worldviews divided only by the vicissitudes of time
.
Macbeth was mismatched with monumental history — wasn’t able
.
Candace Owens
09/22/22
She seemed brilliant at some point
But the lady doth protest too much, me thinks!
https://www.youtube.com/watch?v=ULdJz1EklD0
.
Erin Levy
09/22/22
Mad men S4E5
She made me question my earlier judgments
.
Aphorisms, aphorisms, aphorisms!
.
Mad men has something very American: it’s always about accounts and a partners % contribution to the bottom line
Very, very individualistic even within an organization
Eris! [^31]
[^31] american af
Mad men reconsidered
09/24/22
1. Captures American individualism within the context of a small firm
2. Very much captures the zeitgeist with ERGOTs growth, relocation to NYU, and rechristening
3. My tumultuous attempts to hold forte back at Hopkins
.
My Superpower
09/24/22
1. I don’t judge!
2. Women love this about me
3. And so they share unbelievable shit with me
Beyond Good & Evil?
.
```
Life of Pyrrhus
09/25/22
Pyrrhus was preparing to launch an invasion of the Italian peninsula at great cost to both gold and life. Over a philosophical dinner Cineas asked the king, “Once you have beaten the Romans what shall we do?”
“Once the Romans are conquered we shall have all the riches of Italy at our disposal,” Pyrrhus answered.
Cineas paused, probably sipped from his wine and asked, “And what will we do then?”
Sicily is near! It will be an easy victory.”
Cineas thought a moment more. ”And then what shall we do?”
"Then we will take Libya and Carthage,” the king replied.
"What will we do after that?”
"We will secure all of the Greek world under my rule,” the king nodded at the thought.
“But what will we do then?” Cineas asked.
“Ah, my friend,” said Pyrrhus, ”then we shall rest. We will drink wine, and talk philosophy, and enjoy the fruits of our friendship.”
Cineas looked around. They had wine. They had friendship. They were talking. “Can’t we do what you wish now without harming anyone with war or causing pain to ourselves?”
Pyrrhus’s reply is not recorded. The war went ahead. He died in one of his battles when a tile was thrown from a roof by an enemy woman, smashing his skull. Such accidents rarely happen in dining rooms, so the king would probably have done well to listen to his Epicurean friend and perhaps enjoyed the quieter life of the contemplative thinker.
```
.
Types of sufferers
09/25/22
Known by their challenges:
1. To do (conquer lands, beasts, rule)
2. To be (prepared for final judgement)
3. To hide (in garden when task is conquer)
.
Detraction will not suffer it
.
I’ve had reversals of fortune
09/25/22
And like a phoenix…
.
Swimming
09/25/22
Really enjoyed it yesterday: 10 sets!
.
Mad Men S5E13 : The Phantom
09/26/22
Very relatable, very!
This isn’t fiction
.
RMarkdown [^31]
09/26/22
https://raymondbalise.github.io/Agresti_IntroToCategorical/rmarkdown.html
https://texblog.org/2012/08/29/changing-the-font-size-in-latex/
https://r-charts.com/base-r/line-types/
[^31]: b4 ds4ph
.
That dude has 100 problems!
09/27/22
My new not-so-subtle jab at Rodrigo
.
Kabaniha Grace
09/27/22
Had a wholesome chat with her today
What a joy we deny ourselves!
But what a beautiful day :)
.
Causal inference class
09/27/22
1. Bill Werbel did it
2. Liked it
3. Term 3???
.
Plutarch!
09/27/22
.
Born to…
Rule
Obey
Idler
.
Zeitgeist
09/28/22
Mad men captures my zeitgeist
1. Small firm in large industry
2. Individuals’ contribution to bottom line
3. Disruptions: mergers, acquisitions, loses
This is American at core
```
We can forgive a man for making
a useful thing as long as he
does not admire it.
The only excuse for making
a useless thing is that
one admires it intensely
```
.
09/29/22
The universal language of discourse:
0. Metaphysics
1. Being
2. Knowing
3. Substance
4. Cause
5. Identity
6. Time
7. Space
All men in history may be split into three groups:
1. Time is the essence of everything
2. Seeking truth (i.e., time-invariant essence) is the thing
3. Here and now is the essence and can be accessed by using narcotics & alcohol
Even group membership subject to metaphysics:
1. Changing identity over time, e.g. from 3 -> 2 -> 1 (i.e., becoming)
2. Fixed in identity, i.e., regardless of time (e.g., oh ancient of days!)
3. The issue of which yields most desired outcome
But even desire is subject to metaphysics:
1. Agency (e.g., truthfulness)
2. Ends (e.g., paradise)
3. Passivity (i.e., life) [^32]
[^32]: ape-shit
However, in the end it is a question of “what then”?
1. Doing
2. Being (i.e., truth is best adventure.. Jordan Peterson)
3. Hiding
.
```
*!our
#time e 🤺do it!
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#seeking mc2 ♀ be it
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#now χ ♂ hide
6b.ομορφιά,β
7a.τάξη,α X=0
*!finale
#ooo I 1 1 on 09/29/22
— having ambition/rule
— group or individual fulfillment (i.e., happiness, truth)/obey
— living in the moment/idle
*
— unveiled: willing, changing, becoming, stronger, greater
— veiled: benedict xvi (i.e., anything compromising “the truths” validity)
— everything under the sun has been done, so lets parte after parte after parte
```
.
Finale
09/29/22
1. Time
2. Seeking
3. Now
All invoke “time”:
1. Medium through which their will acts
2. Ultimately irrelevant once “truth” is found
3. Prudence of the suffering man aided by narcotics
.
Back story
09/29/22
1.Two Popes: compelling
2.Mad men: worst infraction
.
GTPCI
09/29/22
1. Big data
2. Health services research
3. AI/Machine learning
4. Translational research
5. Clinical trials
.
Time[^32]
09/29/22
Use it to become harder, better, faster, stronger
Our work is never over
.
```
*!solemn
#time e 🤺doing
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#seeking mc2 ♀ being
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#now χ ♂ hiding
6b.ομορφιά,β
7a.τάξη,α X=0
*!critique
#ooo I 1 on 09/29/22
— post-treatment covariates:
unveiling, i.e., latent phenotypes, becoming
— randomized trials:
truth (veiled in blackbox, i.e., no process)
— meta-analysis of
randomized trials: prudence!
*
— causal inference methods double-down on
variance-models, which is naïve
— neglects epistemological (e.g. latent phenotypes)
& ontological (e.g. sars-cov vars) changes
— everything has been done: so lets just
do a quick meta-analysis of those deeds!
*
— over-time and within an ecosystem agents, verbs & objects
interact, create & destroy ontologies, etc.
— variance in a cross-section of time
(or serial cross-sections over time, varcov structure)
— bored by socratic education, uninspired
by antiquity: humes, fisher, etc.
```
.
```
Survival analysis
09/29/22
Pepe-Flemings is the right p-value for KM survival curves;
log-rank only appropriate if you’ve plotted hazard function
and demonstrated non-cross-over of hazard functions!!!!!!
Ordering of hazards is preserved through integration of hazards
Thus survival functions are also ordered
But going the other way doesn’t imply nicely ordered hazard function
Cumulative hazard may remain ordered yet the hazards cross-over a bit, later on
Here we have the intuition as to why ordered survival may not translate into ordered hazards
So we’ve been abusing the log-rank test in virtually all publications
*
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4626406/
Worth writing a letter to NEJM about this critique
Stat Med is wrong forum!!!
Boy oh boy!
*
The Bayesians have taken over NEJM
My points is moot
Damn!!
```
.
The Economist
09/29/22
Listen to the Liz Truss/Kwarteng article
Some interesting stuff there
Worthwhile
.
Hospitalization
09/29/22
1. Amy’s Table 1 is exciting
2. Consider NIS as control population
3. Preeclampsia? Maybe not conceptually solid
.
```
*!human
#time e 🤺doing
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#seeking mc2 ♀ being
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#now χ ♂ hiding
6b.ομορφιά,β
7a.τάξη,α X=0
*!behavior
#ooo I 2 on 09/30/22
— i am a god
— thou art my god
— atoms, bossons, thats all*
*
— everything has already been done :)
```
.
```
Title of my book
09/30/22
Everything has already been done
1.Start with the epicureans
— Woodstock festival
— Dionysian mysteries
— Nyege Nyege 2022!!!
2.Introduce purpose, mortality
— Religion
— Philosophy
— Culture
3.End with imposing values on others
— Obama
— Trump
— Biden
That’s what an election “mandate is”
But to generalize: that is what life is about
Will-to-power or else live a life of ressentiment
```
.
Iago
09/30/22
I know not if't be true
But I, for mere suspicion in that kind,
Will do as if for surety
.
Tyler Perry
09/30/22
[Revise](https://www.youtube.com/watch?v=fKv0tPJbfSg) my judgement?
.
```
Belgian Ales
09/30/22
#Ale
1.Blonde: Trappiste Westvleteren 95% * Malt (Beginners) ~ Lager
2.Pale: Orval 93%; (Strong): Duvel 94% * Balance: Hops/Malt
3.IPA: Chouffe 93%, Duvel Triple Hop 93% * Hops Mostly
#Trappiste
4.Dubbel: Westmalle 93%
5.Trippel: Van De Garre 97% (+ Unibroue, Westmalle, Karmeliet, Curieux)
6.Quad: Rochefort 10 99% (Westvelteren 100%)
#Sour
7.Oude Gueuze: Tilquin 98%
8.Flanders Red: Rodenbach 99%
9.Lambic: Oude Lambik 3 Fonteinen 91%
*
Where does Saison fit in this scheme?
```
.
```
*!critical
#time e 🤺doing
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#seeking mc2 ♀ being
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#now χ ♂ hiding
6b.ομορφιά,β
7a.τάξη,α X=0
*!race theory
#fwn II 2 2 on 09/30/22
— i am a god: monumental
— thou art my god: antiquarian
— bosons, that’s the force: critical
*
— tyler perry, oprah winfrey, king richard, earl woods are refutations
— conservatives only want to talk about greatness of our country
— proponents are critical of this single-perspective of history
*
— doing, willing, becoming, stronger, harder, faster, better.. our work is never over
— seeking the pathway to paradise, making the right decisions, staying true
— everything has already been done: big-bang, super-symmetry, whatever
```
as of 06/2023 i've [swapped](https://muzaale.github.io/book/bilowozo.html#time) this as follows:
1. monumental
2. critical
3. antiquarian
.
Zeitgeist
09/30/22
Word of the month
1. Mad men
2. D’Mile
3. Lucky Daye
4. Smokin’ out window
5. Don Draper
.
```
*!zeitgeist
#time e 🤺doing
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#seeking mc2 ♀ being
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#now χ ♂ hiding
6b.ομορφιά,β
7a.τάξη,α X=0
*!creative destruction
#fwn II 2 2 on 09/30/22
— zeitgeist
— volksgeist
— weltgest
```
.
Four score and seven years ago…
09/30/22
I can’t breath 2:22/4:46
1. H.E.R.
2. D’Mile
3. Tiara Thomas
https://www.youtube.com/watch?app=desktop&v=E-1Bf_XWaPE
#### 10/2022
Bob Dylan
09/30/22
Your money meant shall be my gentle verse,
Which eyes not yet created shall o’er read;
And tongues to be, your being shall rehearse,
When all the breathers of this world are dead;
You still shall live,— such virtue hath my pen,—
Where breath most breathes, even in the mouths of men.
Review Bob Dylan’s acceptance speech
For the Nobel prize in literature
https://www.nobelprize.org/prizes/literature/2016/dylan/lecture/
.
Reversal of fortunes
10/01/22
Idea of the month
.
Swolf is a score that measures your swimming efficiency, and just as in golf, a lower score is better. For pool swimming, the swolf score for a length is obtained by adding together the number of strokes taken in the pool length, and the time it took to swim that length.
.
```
Avg. SWOLF
10/01/22
#25 Butterfly 25yd 0’48”/28
Avg.SWOLF 19
Lowest on record!
#34 Butterfly 25yd 0’57”/20
Avg.SWOLF 19
#46 Butterfly 25yd 0’43”/28
Avg.SWOLF 17
*
Swolf is a score that measures your swimming efficiency, and just as in golf, a lower score is better. For pool swimming, the swolf score for a length is obtained by adding together the number of strokes taken in the pool length, and the time it took to swim that length.
*
<35 for long axis
<25 for short axis
```
.
https://www.youtube.com/watch?v=N8av28fydMw
.
It’s my theory that men who drink *strictly* one alcohol beverage (Guinness in this case) have so much trouble sampling strictly one chic. Mwami Tumwiine, aka Isabirye :)
10/01/22
16:50hrs
.
Creative destruction
10/02/22
1. Symptom of energy
2. Consequence of becoming
3. Especially while standing on shoulders..
4. Monumental historians with dynamite!
5. They see beautiful bricks, not the building
.
Interesting
10/02/22
1. Long legs, peak-to-peak, perspective, effects!
2. Navel-gazing, causes, no mentors!!!
3. Everything has been done
.
Stata Programming
10/03/22
1. Blocks of functional code
2. Analogous to genes
3. Include regulator “if” statements
4. Thus “if 0” vs. “if 1” vs. “if 2” vs. “quietly”
5. Would regulate specific bits of the script/code
.
Porter, Stout, Coffee, Cocoa
10/03/22
All based on roasted seeds
Might be blended together in beer
Generally darker than unroasted stuff
.
```
Mary Grace
10/03/22
Precision Medicine Analytics Platform (PMAP) data
— huge new asset!!!!
— cause for delay in IRB?
— maybe bypass
Interested in using it? Reach out to MGB
*
CVD following KT
Leading cause of death
Excluding pre-KT CVD??
*
https://pm.jh.edu
*
Build database as standard of care
Feed it back into EHR dashboard
Has already been done by biostats department
*
Submit IRB &
Add to MGB IRB???
Candidates, Recipients, and their donors!!!
MGB may help out here since its expensive
Delivered in a data projection on SAFE desktop
eForm R (Christine’s) —> for IRB
eForm S (MGBs) —> supplement of eForm R (from Christine)
Would potentially use eForm S on MGBs grant
```
.
Matrices
10/04/22
```r
H=X %*% solve(t(X) %*% X) %*% t(X)
#age 65
diff65 = coefficients(fit_2.1)[5]/1000 se65 = sqrt(vcov(fit_2.1)[5,5])/1000 CI65 = diff65 + c(-1,1)*1.96*se65
#age 75
A = c(0,0,0,0,1,10,0,0)diff75 = as.numeric(t(A)%*% coefficients(fit_2.1))/1000 se75 = sqrt(as.numeric(t(A)%*%vcov(fit_2.1)%*%A))/1000 CI75 = diff75 + c(-1,1)*1.96*se75
```
.
Madness in great ones must not unwatched go
10/05/22
https://www.psychiatrictimes.com/view/shakespeare-and-psychiatry-personal-meditation
.
What is a man? If he foreseeth evil what does he? Hideth? Whineth? Or Tameth?
10/05/22
My wittiest aphorism
Aphorism contra Proverbs 27:12 contra Psalm 23
.
In lieu of TV:
10/05/22
https://www.youtube.com/watch?v=GO0Pzh3BFMk
https://www.youtube.com/watch?v=0bKX5quiYhU
.
Counterfeit
10/05/22
Word of the month
Best word of all-time
Summarizes everything!
.
Brazilian Gourmet
10/05/22
1. Burger
2. Pao de Quejo
3. Coxhinha
4. Bolhinho de …
5.
my #1 ethnic food from S.A.
.
Yale
10/05/22
English department
http://www.curtainup.com/richard2yale07.html
god save the king! will no man say amen?
A CurtainUp Review
Richard II
By Summer Banks
.
Shakespeare & Modernity
10/05/22
1. Absurdity/Satire
2. Trial of Hamlet/C-SPAN
https://www.youtube.com/watch?v=GO0Pzh3BFMk
https://www.c-span.org/video/?55363-1/trial-hamlet
.
NHATS
10/05/22
— Medicare beneficiaries
— Longitudinal data
— Insights
— Foot prints of missing data
— Planning live donor data collection revisions
.
```
Thesis Committee
10/05/22
1.GTPCI advisor/Julie (jhoover2@jhmi.edu)
2.SOM thesis mentor/Dorry (dorry@jhmi.edu)
3.Another GTPCI/Betsy (eking19@jhmi.edu)
4.Primary SPH/Caffo (Chair) (bcaffo1@jhu.edu)
*
1.GPTCI/Fabian (fjohnst4@jhmi.edu)(newkir3@jhmi.edu)
2.Primary SPH/Alden (agross14@jhu.edu)
```
1. Healthy older non-donors
2. Incidence of hospitalizations (ICD-9/10)
3. Trajectories of frailty
4. Cross-sections of donor hospitalizations
5. Footprint of missing data: see Macey
.
```
Jen Schrack
10/06/22
*by looking at NHANES we’ve revealed that donors, though healthier than average, have increased risk —> AR
*first mortality, then ESRD, now we wish to go further upstream before ESRD (e.g. CKD, CVD, hospitalization, frailty)
*with older NHANES we don’t have great longitudinal data for our upstream interests
*here’s where NHATS comes to mind with caveats…
*there is no longitudinal older donor followup. At best cross-sections during hospitalization events…
1.Healthy older non-donors
2.Incidence of hospitalizations (ICD-9/10)
3.Trajectories of frailty
4.Cross-sections of donor hospitalizations
5.Footprint of missing data: see Macey
In that case, let’s meet in my office at COAH: 2024 E Monument St, Suite 2-700, room 710. See you then!
— disability trajectories: ADL, IADL, frailty, etc.
— cms-linked restricted file for ESRD: my access? IRB,
— incidence of hospitalizations and associated ICD codes!!!!
*
— guide my cross-sectional donor analyses
— maybe cross-sectional comparisons
— unlikely to have disability collected in donors
— but will use text review of medical records… length of hospitalization, rehospitalization, etc.
*
— also pilots for R03/R21?
*
Physical, cognitive functional trajectories, but not blood work…
2 and half hours at home…
SBBP, cognitive battery, hearing and vision, etc.
Hospitalization, CMS (free linkage).. next c
ACJMEN
*demographicas can be linked with medical claims
2011-onward
Replenished in 2015
Covid mesed 2020 replenishment
1.website… instructions
2.application to fill out
3.DUA with Hopkins/HATS/NIA (two diff. Applications)
4.IRB
5.NIA approval
https://www.nhats.org
The unclave…
*
The other study—> ARIC… may have a lot of measure too.
Go to the website… fill out a manuscript proposal form… would help with labs
```
.
Criminal responsibility
10/06/22
1.Sanity or insanity
2.Right before the criminal act
3.Does premeditation invalidate time immediately prior?
https://www.c-span.org/video/?55363-1/trial-hamlet
Timeline is everything
Bipolar, rapid cycling for instance
States come and go: being (truth) vs. becoming (process)
33:15/2:09:36
.
```
Time
10/06/22
1.Time implies several interacting processes
2.Q&A seeks some invariant being
3.Mutually exclusive: 1 vs. 2
— philosophy: parmenides/einstein vs. heraclitus/schroedinger
— competing risks: latent distribution if reality hadn’t happened!
— religion: was, is, ever shall be.. has no changeable “mind” like man
— science: isotopic only if viewed at the physically impossible speed of light!
— justice: premeditation (being) vs. immediately before action (process)
— diagnosis: rapidly-cycling bipolar disorder, manic now, depr..
```
.
Counterfeit
10/7/22
What art thou. That counterfeit'st the person of a king?
The knave counterfeits well. A good knave
I fear thou art another counterfeit,
Now counterfeit to swoon, why, now fall down
.
On schedule today
10/08/22
1. Patton
2. Hamlet
3. Lebowski
No more after this!
.
Wedding anniversary
10/08/22
Nov 17, 1972
.
Kunswaaza
10/08/22
The Road to Kirya
Maurice Kirya
.
Alejandro G. Iñarritu Still Hurt by Robert Downey Jr.’s
Response to His Belief That Superhero Films
Are ‘Cultural Genocide’
09/08/22
Alejandro Gonzalez Iñarritu does not like superhero movies.
The Oscar winner said in 2014 while promoting
“Birdman” that superhero movies were a form of
“cultural genocide,” adding, “I don’t respond to those
characters. They have been poison because the audience
is so overexposed to plot and explosions and shit that
doesn’t mean nothing about the experience of being human.”
10/10/22
Tameth/Malcontent (Innate)
Whineth/Jesus (Source)
Hideth/Police (Prefrail)
Hope of deliverance from without!
At least religion’s error results
In cultural ties and bonds :)
21 grams? Del Toro!!!
.
```
*!fave
#will e
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ
#metaphysical mc2
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ
5c.φάρμακον,δ
#physical χ
6b.ομορφιά,β
7a.τάξη,α
*!auteurs
#adm V 6 on 09/03/22
— agency, dynamite —> atoms //quentin tarantino
— cause —> effect —> forever //woody Allen
— passivity, atoms —> narcotics //coen brothers
*
bowling, ransom, nihilists, ruined rug
*
— monumental
— antiquarian
— critical
#ooo take II 10/10/22
— tameth/rigged: alejandro.iñarritu
— whineth/scream: coen brothers
— hideth/banter: woody allen
```
.
Takes a Village
10/10/22
1. Spartans raised their children in cohorts, not families
2. British-style boarding school does the same, at Eton
3. The whole family-values argument is flawed, Duncan!
.
Butiful
10/10/22
My love, what you see
Over there aren’t the stars:
It’s your nervous system
Alejandro G. Iñarritu
.
Iñárritu has stated that he believes music has had a bigger influence on him as an artist than film itself
.
Innaritu
https://www.youtube.com/watch?v=E2xuTyILwTw
.
Amores Perros
10/12/22
1. Sparta: soldiers or workers
2. Britain: boarding school
3. Guadalajara? a finger up his ass!
Family values my ass!
```
Know what? A baby born in Guadalajara
gets a finger up his ass.
- Why? - To see what he'll be.
If he kicks, he'll be a soccer player.
If he screams, a mariachi.
And if he laughs, he'll be a homo.
What about girls?
We finger them until they're 18
```
.
Sparta was frequently a subject of fascination in its own day, as well as in Western culture following the revival of classical learning. The admiration of Sparta is known as Laconophilia. Bertrand Russell wrote:
Sparta had a double effect on Greek thought: through the reality, and through the myth.... The reality enabled the Spartans to defeat Athens in war; the myth influenced Plato's political theory, and that of countless subsequent writers.... [The] ideals that it favors had a great part in framing the doctrines of Rousseau, Nietzsche, and National Socialism
.
Duncan?
10/12/22
Spartan boys were expected to take an older male mentor, usually an unmarried young man. According to some sources, the older man was expected to function as a kind of substitute father and role model to his junior partner; however, others believe it was reasonably certain that they had sexual relations (the exact nature of Spartan pederasty is not entirely clear). Xenophon, an admirer of the Spartan educational system whose sons attended the agoge, explicitly denies the sexual nature of the relationship.
.
```
*!λακωνίζειν
#perros e 🤺
0g.κοσμογονία,γ
1f.ἡ ἔρις,κ
2e.πρᾶξις,σ r2
#amores mc2 ♀
3e.ἄσκησις,μ
4d.∫δυσφορία.dt,ψ #i.
5c.φάρμακον,δ Ho
#dolor χ ♂
6b.ομορφιά,β
7a.τάξη,α X=0
*!laconophilia
#ooo take III on 10/12/22
— monuments sparta
— antiques britain
— critique guadalajara
*
— monarchy/tyranny/deus
— aristocracy/oligarchy/telos
— democracy/anarchy/chaos
*
— agoge system
— boarding school
— finger up his ass
```
.
Critical Race Theory :)
10/12/22
https://www.youtube.com/watch?v=_xQ27AsF4_k
.
Iñárritu
10/12/22
Finds his ἀγών in Birdman
.
Why do they call the Ukraine war “unprovoked”?
It’s been known since the beginning of time that nothing like weakness provokes the strong!
They don’t treat you like a worthy adversary but an easy addition to their labor force
.
Life
10/13/22
It’s all about knowing when to tame, whine, or hide:
Ecclesiastics 3:1-8
.
Abimelech was the generic name given to all Philistine kings in the Hebrew Bible from the time of Abraham through King David. In the Book of Judges, Abimelech, son of Gideon, of the Tribe of Manasseh, is proclaimed king of Shechem after the death of his father. Wikipedia
.
Calvin
10/14/22
1. Outstanding job!
— Table looks tidy
— Statistics believable
— But its not yet perfect
2. New concepts
— Inclusion criteria
— Recoding variables
— Merge vs. append
3. Table1_Nondonors
— Overall n=76,153
— Exact grouping required (e.g. Race: White, Black, Hispanic, Other)
— Don’t include any subgroups that are not shown in Table_Nondonors (.e.g. Citizenship)
.
10/08/22
gamma function and exponential distributions
these are dominated my mind
https://en.wikipedia.org/wiki/Gamma_function
counterfeiting data even so!
```stata
//swoosh
cls
clear
local n=10^4
set obs `n'
g beta=_n
replace beta=beta*7.3/`n'
g fb = exp(lngamma(beta))
g logfb = lngamma(beta)
line logfb beta, sort
graph export swoosh.png, replace
```

.
Swinging between genius and idiocy… has an attention span of 3 seconds
.
Drugs still sellin
Snitches still tellin
.
As I Like It
10/16/22
I want people in places at certain times
Acting like people in places at certain times
Not some meta-analysis of such people, places, times
.
Donald Glover, I’ve written you off!
.
De devil hideth in de tails of de distribution
10/18/22
Meanwhile God reigneth in the middle and tameth it to his will
As such, the predictions and forecasts that come forth from parametrization are of questionable if any value to the sort who will not be governed: heroes and great men, on the one hand, and slackers and epicureans on the other
.
Ted Lasso
10/18/22
Only such a broken man could write so moving a TV show!
— Nanny diaries
— Saladgate
— Etc.
From harmony within he wrung out order, harmony, and beauty in a world gone awry. This is aesthetic
.
Lucy Fa vs. De Telos
10/18/22
War of the tails :)
.
St. Paul
10/19/22
I propose that Dr. Kasenene rebrand as St. Paul
.
```
Sudeikis
10/19/22
#fwn III 2 201
Wit.—Wit is the epitaph of an emotion.
— mythos: ted lasso as a tragicomedy in finest form
— logos: q&a, capitulating to art, bravo!
— pathos: auteurs divorce
*
— from harmony within
— wrung out order, beauty in an awry world
— nanny, saladgate, harry styles, quite [olivia] wilde innit?
```
.
My Synonyms
10/18/22 - present
I
Superman, mythos, argos, ongoing, monumental, aristophanes, will, idiosyncrasy, delivereth, generosity, y, perros, action, tournaments, alone, hades, zeus, olympus, eagle/self, domination, sanguine, tyranny, heraclitus, desert, aphorisms, bge, lordstar, god, judges, eternal recurrence, tomorrow and tomorrow and tomorrow, figs, wild honey, locusts, festivals, psilocybin, soul, energy, nietzsche, power, iñárritu,
II
God, logos, telos, story, antiquarian, socrates, theory of everything, parameters, tameth, order, β, amores, verb, busy, cult, clouds for floor, stars for chandelier, heaven, church/superego, missions, the blood, monarchy, parmenides, paradise, beatitudes, good, priest, leader, romans, redemption of man, forever, bread, wine, sacraments, incense, spirit, chastity, freud, stunted, perry,
III
Devil, pathos, narcos, backstory, critical, epicurus, atomos, tvar, hideth, chaos, X, dolores, object, passive, herd, green pastures, still waters, america/id, hopkins, grants, belgians, anarchy, democritus, garden, eden, original sin, evil, satan, tail, genesis, fall of man, now, food, drugs, cigarettes, alcohol, sex, marx, weak, allen,
.
PYt :)
10/20/22
— survival —> Area: P(Y < t = T or C)
— event —> Limit: C
— time —> X: T
.
— P(T>t, C>t) = S(t)
— Y=min{T,C}; Delta=I(T<C)
— t
.
— here do simulation
— assume pars
— generate ts
.
```
Migration
10/21/22
1.Migrate from notes to RMarkdown
2.Strict format (see my.pyt.Rmd)
3.Hyperlinks, color codes!
```
.
Maldives
10/22/22
Dinye Mawanda
Angela Aizire
Bunda Katende
Leona Buhenzire
.
Westmalle
10/22/22
After a seven-year itch
I conclude that this is my #1
Belgian across all styles
.
Vanessa Carlton
10/22/22
https://www.youtube.com/watch?v=Cwkej79U3ek
707k subscribers
335m viewers :)
.
```
Bryan Adams
10/22/22
Let’s make a night to remember 2:55/4:33
Have you ever really loved a woman? Hahahaha
Bryan Adams has been a vegan for 32 years; he quit eating meat and dairy in 1989.[352] He relayed his experiences with his plant-based diet in an interview with Vegan Life Magazine in 2016:
For those people who aren't veggie or vegan it was the best gift I could ever give myself to do it. I am turning 57 years old this year and I work hard, I am always on the move but I have tons of energy because I am plant-based. It is absolutely the best thing you could ever do for yourself. It is a great path.[353]
Adams has never married. In the 1990s, he was in a relationship with Danish model Cecilie Thomsen.[354] Adams and Alicia Grimaldi, his former personal assistant and now trustee and co-founder of his namesake foundation, had their first daughter in April 2011 and their second daughter in February 2013.[355] His parents are British, and one grandmother was born in Malta.[356] Adams has homes in London[20] and Paris.[357]
On October 30, 2021, Adams cancelled his participation in a tribute to singer Tina Turner, just before the Rock and Roll Hall of Fame ceremony, after testing positive for COVID-19.[358] On November 25, 2021, Adams tested positive for COVID-19 for the second time, and was placed in a hotel for a two week quarantine in Italy
```
.
Rottweiler
10/22/22
I’ve a soft spot for this breed
.
Guillermo
10/22/22
https://en.wikipedia.org/wiki/Guillermo_del_Toro
.
```
Auteurs
10/23/22
1.Alejandro Iñárritu
2.Alfonso Cuaron
3.
These motherfuckers don’t offer any metaphysical consolation
Maestro Cuaron might appear to do so in Sólo con tu pareja
But the final scene is really a super-subtle critique of consolation
*
As for sampling the “distribution” they do a nice job
Each of the strata is represented
Mean, both tails, etc.
*
Random variables?
Persons, places, times
Under persons: race, ethnicity
*
What about places: often auteurs hometown
Of course there is room for “away”
For places: past epoch, current one/never future!!!
```
.
Amazing how business schools are still stuck with case-study method in the day and age of science. Instead of these hallowed HBS case studies, folks should have access to data from teams across the world and rigorously analyze it to find patterns
.
Rishi (to recap whom they passed over when they picked Liz Truss:)
1. Winchester (like SMACK)
2. Oxford
3. Stanford
4. Goldman Sachs
5. Hedge Fund
6. Chancellor
7. Resignation ended Boris Johnson
& Married an Indian chic
Father-in-law is a billionaire
.
```
GDP
1.US $19T
2.China $12T
3.Japan $5T
4.Germany $4T
5.India $3T
```
.
Uganda is statistically significantly poor with a GDP of 0.03% of the world
This is an error in logic since it’s the same contribution as Estonia
However, Estonia achieves this with a population of 1.3m vs 41.1m for Uganda
.
[LaTeX](https://wjgan.com/posts/latex.html)[^34]
[^34]: LaTeX
.
Yield spread
10/25/22
https://fred.stlouisfed.org/series/T10Y2Y
1. downlaod dataset
2. read into RStudio
3. include hyperlinik
.
Intuition
10/25/22
Second derivative of log function
And statistics
Variance?
With MLE of theta
The variance is - d2/d theta^2
https://data.princeton.edu/wws509/notes/c7s2
.
Fisher information!
.
Passive voice
At 1:03/6:03 on 10/26/22
And [I](https://en.wikipedia.org/wiki/Rishi_Sunak) want to pay tribute to my predecessor, Liz Truss.
She was not wrong to want to improve growth in this country.
It is a noble aim. And I admired her restlessness to create change.
But some [mistakes were made](https://en.wikipedia.org/wiki/Passive_voice). Not bourn of ill will or bad intention:
Quite the opposite, in fact. But [mistakes, nonetheless](https://twitter.com/RishiSunak/status/1584892345715040258?ref_src=twsrc%5Egoogle%7Ctwcamp%5Eserp%7Ctwgr%5Etweet).
.
```
Abimereki Dhatemwa at 11:24 on 10/26/22
Unfortunately, these platitudes are meaningless
They’ll not make a convert of this fella ☝🏾
Uniting the country? That’s what I was reacting to. Folks from larger previously dominant tribes are threatened by unity. Things can only get “worse” for them as the “United Nations” take their country from them
We see it with the Baganda and Wazungu in America, UK, Sweden, Spain, Italy, France, etc
Also, and this is a question for P, as father, Christian, and educator: when one talks of family values, education, and raising our children, aren’t all these things at war with each other?
I say this as a product of all the above:
1.Church of Uganda did its job in childhood
2.Boarding school did its job in teenage
3.Wider world has done its job in early adulthood
4.And family has been the accompanying bass through out
5.Then science, philosophy, arts have proved to be disrupters of everything that’s I’ve been taught since childhood as “valuable”
So to me family values compete with education. They don’t complement each other. It’s the whole mess of boarding school and the various influences it brings. Is it the point to shield onself from outside influence? If so, then why not just remain at home instead of school?
Food for thought
And Ps responses especially desired since he is national leader in education, ministry, family, counseling :)
One last thing, with the ascendency of Indian culture in the UK (curry, kebab now not Fish & chicken and plum pudding), and the already eminent decline of the Church of England, Hindu May emerge as a source of values
Is a Christian to welcome these changes and trends?
This is the world my nieces and nephews are going to face as they seek further education and career opportunities beyond Kampala
*
Patrick Muinda at 15:32 on 10/26/22
D, thanks for asking.
When done as God teaches us in His Word, family values and Education actually complement each other. They are by no means at war with each other when done as taught by God in His Word.
Let's examine the facts in scripture:
Exodus 20:12 (The First family value, given as a command AND the only command in scripture with a promise)
Regard (treat with honor, due obedience, and courtesy) your father and mother, that your days may be long in the land the Lord your God gives you. A promise of long life!! ...as a desirable outcome of family values. Education alone cannot pull off long life. We've seen Proffessors cut their lives short by simply not living according to God'sWord.
2 Timothy 3:15 says, you took in the sacred Scriptures with your mother's milk as she taught you Godly, damily values! There's nothing like the written Word of God for showing you the way to salvation, and success in this life through faith in Christ Jesus. (...added emphasis).
This too applies to formal education. As you study scripture to add to your knowledge of God and His will, we too ought to have a formal education to add to our knowledge of life in this world.
Jesus prayed for us when He said in John 17:15-16 My prayer is not that you take them out of the world but that you protect them from the evil one. They are not of the world, even as I am not of it.
On the flip side, having Godly family values will keep you away from evil. Education does not even attempt to do likewise. If anything, education could draw you into science that instead draws you away from God.
The Apostle Peter exhorted believers to “make every effort to add to your faith goodness; and to goodness, knowledge, and to knowledge, self control” (2 Peter 1:6).
When you couple this with the family values enshrined in honoring your father and mother, it will go well with you in the land God has given you...even your education and it's outcome thereof.
Finally, being a wholesome Christian who is educated and yet is established in Godly family values, the outcome is the creation of an environment of respect for God, for His Word and for authority. It will lay foundations of discipline, moral values and a work ethic that will prepare a student for life. That student will grow into such a pullar in society because He is well anchored in God's Word with education and solid family values.
I submit. God be with you.
P.
```
.
Let us hear the conclusion of the whole matter: Fear God, and keep his commandments: for this is the whole duty of man
.
Oktoberfest: Blood & Beer
It’s so cutthroat !!
.
#### 11/2022
Should we spend our day, like herds, grazing upon the green pastures and the still waters that we beg of our shepherd? Don’t have much loftier tasks? Like going afield to conquer other terrains, other Nietzsche’s.. including desert for oil…
11/01/22
So where do you think the strongest animal - the gorilla - gets it’s protein to build all that muscle?
Where do cow’s and goats get protein to give us?
A riddle that meat = protein enthusiasts can’t solve
.
Societies that eat little meat live longest?
Italy and Japan???
.
```
Thesis advisory committee
11/01/22
1.Approved research plan
— why over 2-4y period?
— several reasons
— considered original timeline too ambitious (both GTPCI and NIA reviewers!)
— following recommendations, I extended out my plans for an additional 2 years
2. Since then
— scheduled and passed my PoE
— have secured most of the data I’ll need (corresponding to 1-3)
— my immediate next steps are curating analytic datasets
— exploratory analyses and reporting preliminary findings
— iterations and feedback from you
3.Goal is to be at stage for thesis draft, defense, and publication by may/2023
— ambitious as advisory committee says
— but will give it a try
— still have some advanced courses to complete
— one in each term..
4.any questions???
— thesis research documentation
— due academic registers office on 11/26
— finalize this in next two weeks
—> Fabian not available in first week of May 2023
Hi Fabian,
Looking forward to a brief discussion of my thesis plans with you and two other advisory members. I’ve attached a brief outline of what I presented for my preliminary oral exam. And have also attached the approved research plan.
Here’s my update:
1. Research plan & timeline approved by the GTPCI advisory committee
2. Since approval I’ve completed 1-3 of 9 tasks
3. On course to complete 4-6 over next two months
4. Next scheduled Thesis advisory committee meeting ~ 05/23
5. Aiming to have completed 1-6 and possibly 7 by then
Thanks,
Abi
1.
Fabian Johnston fjohnst4@jhmi.edu
Trashawnna Hollingsworth thollin4@jhmi.edu
Julie Hoover-Fong
Dorry
```
.
```
Synonyms
Personal god
Dionysus
I will
Autonomy
Independence
Mythos
Vs
Herd animality
Apollo
Thou shalt
Vulnerability
Dependency
Logos=Ethos
```
.
Jesus does everything but score: He often has the facial expression of someone who just lost their winning lottery ticket and that tells you how desperate Gabriel Jesus is to succeed
.
```
Bill Werbel
11/07/22
Building a “Platform” Cohort Study
— data structure
— institutional memory
— so that recycling thru TRC doesn’t cause trouble
— NHANES, anybody??
Diane Brown
Database and data management initiative: standardization of the TRC databases design process and data management plans
— this is some really cool stuff
— data management plan for all NIH grants in 2023
— PMAP??
— overarching IRB the covers all solid organ transplant!!!
— did she just add me to the IRB?
```
.
Fillibaster
11/08/22
```r
p=.51; n=100; se=sqrt(p*(1-p)/n); p; p+c(-1,1)*1.96*se
```
The filibuster is a powerful legislative device in the United States Senate. Senate rules permit a senator or senators to speak for as long as they wish and on any topic they choose, unless "three-fifths of the Senators duly chosen and sworn"[54] (usually 60 out of 100 senators) bring debate to a close by invoking cloture under Senate Rule XXI
Incidentally, 3/5 (or 60/100) is the upper bound of the 95% confidence interval of the smallest possible majority, 51%. Coincidence?
.
$
[\prod_{i=1}^n]
$
.
**Yours truly:** convert this R code to python
**ChatGPT:** Certainly! Here's the equivalent Python code for the given R code:
<code>
import math
p = 0.51
n = 100
se = math.sqrt(p * (1 - p) / n)
confidence_interval = [p + x * 1.96 * se for x in (-1, 1)]
print(p)
print(confidence_interval)
</code>
This Python code calculates the standard error (se) using the formula sqrt(p * (1 - p) / n), where p is the probability and n is the sample size. It then calculates the confidence interval by adding and subtracting 1.96 * se from the estimated probability p. Finally, it prints the probability p and the confidence interval.
```
Ecclesiastics 2
11/09/22
https://www.biblegateway.com/passage/?search=Ecclesiastes%202&version=KJV
*
Fully of vexation come I
http://shakespeare.mit.edu/midsummer/midsummer.1.1.html
Full of vexation come I, with complaint
Against my child, my daughter Hermia.
Stand forth, Demetrius. My noble lord,
This man hath my consent to marry her.
Stand forth, Lysander: and my gracious duke,
This man hath bewitch'd the bosom of my child;
Thou, thou, Lysander, thou hast given her rhymes,
And interchanged love-tokens with my child:
Thou hast by moonlight at her window sung,
With feigning voice verses of feigning love,
And stolen the impression of her fantasy
With bracelets of thy hair, rings, gawds, conceits,
Knacks, trifles, nosegays, sweetmeats, messengers
Of strong prevailment in unharden'd youth:
With cunning hast thou filch'd my daughter's heart,
Turn'd her obedience, which is due to me,
To stubborn harshness: and, my gracious duke,
Be it so she; will not here before your grace
Consent to marry with Demetrius,
I beg the ancient privilege of Athens,
As she is mine, I may dispose of her:
Which shall be either to this gentleman
Or to her death, according to our law
Immediately provided in that case.
```
.
Senate/Congress
11/09/22
```r
p=.51; n=100; se=sqrt(p*(1-p)/n); p; se; p+c(-1,1)*1.96*se
p=.51; n=435; se=sqrt(p*(1-p)/n); p; se; p+c(-1,1)*1.96*se
```
<code>
import math
# given variables
p = 0.51
n = 100
# calculate standard error (se)
se = math.sqrt(p * (1 - p) / n)
# calculate the 95% confidence interval
ci_low = p - 1.96 * se
ci_high = p + 1.96 * se
print(f"p: {p}")
print(f"se: {se}")
print(f"95% confidence interval: ({ci_low}, {ci_high})")
</code>
<code>
# update n
n = 435
# calculate standard error (se) with the new n
se = math.sqrt(p * (1 - p) / n)
# calculate the 95% confidence interval with the new se
ci_low = p - 1.96 * se
ci_high = p + 1.96 * se
print(f"p: {p}")
print(f"se: {se}")
print(f"95% confidence interval: ({ci_low}, {ci_high})")
</code>
**Yours truly:** reasonable null hypothesis for a philibaster in senate?
**ChatGPT:**
Apologies for the confusion, but there seems to be a misunderstanding. The provided Python code correctly calculates a 95% confidence interval around a proportion, and the output should not exceed the range of 0 to 1, as a proportion is a percentage and cannot exceed 100% or be less than 0%.
When you calculate the standard error and the confidence interval with the given values (p=0.51, n=100), the result will fall within this range. If you're obtaining a value of 60, it suggests a potential issue with the calculation or an error in the code's execution.
Let's do a quick check on the calculation:
```
```python
import math
# given variables
p = 0.51
n = 100
# calculate standard error (se)
se = math.sqrt(p * (1 - p) / n)
# calculate the 95% confidence interval
ci_low = p - 1.96 * se
ci_high = p + 1.96 * se
print(f"p: {p}")
print(f"se: {se}")
print(f"95% confidence interval: ({ci_low}, {ci_high})")
```
With the above code, the output should be something similar to:
```yaml
p: 0.51
se: 0.04998999899979995
95% confidence interval: (0.41202664796364, 0.60797335203636)
```
The confidence interval falls between roughly 0.41 and 0.61, which are reasonable bounds for a confidence interval around the proportion 0.51. This is consistent with the expected range (0-1) for a proportion.
**Yours truly:** so for the US senate that has 100 members (n=100), doesn't a null hypothesis (p=.51 for a razor-thin senate majority) give us an upper bound of 60? Which is the philibaster number?
**Yours truly:** Apologies for the earlier confusion, but I understand your point now. You are not looking at the proportion as a percentage between 0 and 1, but you're looking at it as the actual number of senators (out of 100). When you calculate the upper bound of the 95% confidence interval with p=0.51 and n=100, you're looking at the potential number of senators who might support a bill, not a percentage.
**ChatGPT:** If we revisit the calculations:
```python
import math
# given variables
p = 0.51 # proportion of senators
n = 100 # total number of senators
# calculate standard error (se)
se = math.sqrt(p * (1 - p) / n)
# calculate the 95% confidence interval
ci_low = n * (p - 1.96 * se) # lower bound of the interval (number of senators)
ci_high = n * (p + 1.96 * se) # upper bound of the interval (number of senators)
print(f"p: {p * n}")
print(f"se: {se * n}")
print(f"95% confidence interval (number of senators): ({ci_low}, {ci_high})")
```
This will give you the range of senators (out of 100) who might support a bill with 95% confidence. The upper bound should indeed be around 60 senators, which is the threshold number for breaking a filibuster in the Senate.
Note: We're assuming here that the true proportion of supporting senators in the population is 0.51, and we're calculating a confidence interval around that proportion. The calculation doesn't directly address the chance of a filibuster or its success. It's just a measure of statistical uncertainty around the estimate of supporting senators.
**Yours truly:** any evidence that this is the logic behind the "magical" 60?
**ChatGPT:** The "magical" 60, as it relates to the United States Senate, refers to the number of votes required to invoke cloture and end a filibuster, or extended debate that blocks legislative action. This number is not directly linked to statistical analyses or confidence intervals, but instead, it is a procedural rule set by the Senate.
The reason why 60 votes are needed to end a filibuster is due to Rule XXII of the Senate, often called the "cloture rule". This rule was adopted in 1917 in response to the tactics of obstructionists, particularly during debate over America's entry into World War I. Originally, two-thirds of senators present and voting were required to invoke cloture; this was changed to three-fifths of all senators (normally 60 out of 100) in a 1975 amendment to the rule.
In practice, the 60 vote rule means that, while a simple majority (51 votes) is required to pass most legislation, a supermajority (60 votes) is required to end debate and bring the legislation to a vote. This gives substantial power to minority factions within the Senate, as they can block legislation unless a compromise can be reached that convinces enough senators to invoke cloture.
While it's an interesting observation that the upper range of a 95% confidence interval around a slim majority (0.51) in a 100-member Senate is near 60, it's likely coincidental and not the direct reasoning behind the procedural rule. The rule is more about facilitating debate and requiring a larger consensus to move forward with legislation.
.
eIRB JHU
11/09/22
Do it!!
.
```
Lack of rigor: recommendations for improvement
— Choose retracted article for part A
O-Primary prevention of cardiovascular disease with a Mediterranean diet
E-Did the PREDIMED Trial Test a Mediterranean Diet?
R- Published
0.mu/expected - f(y) /observed = error
1.I like random errors
2.Redundacy design (e.g. meta-analysis) address this
— Then do part B
https://www.hsph.harvard.edu/nutritionsource/2018/06/22/predimed-retraction-republication/
On June 13, 2018, the New England Journal of Medicine (NEJM) retracted the 2013 study, “Primary Prevention of Cardiovascular Diseases with a Mediterranean Diet
*
PART A
Estruch et al for the PREDIMED Study Investigators. Primary prevention of cardiovascular disease with a Mediterranean diet. NEJM 2013
A systematic review and meta-analysis of 5087 randomized, controlled trials published in general medical journals was conducted by a meticulous defender of science, Carlisle JB, in 2017 (1). Carlislse found data fabrication and other reasons for non-random sampling in several of these studies, including Estruch et al for the PREDIMED Study Investigators. Primary prevention of cardiovascular disease with a Mediterranean diet. NEJM 2013 (2). This latter study is my choice for Part A of this weeks activity.
Table 1 in this very highly cited study demonstrated differences between baseline study and control populations characteristics that were substantially different from what would have been expected to result from randomization. A year later in 2018, the study authors in collaboration with the NEJM editors retracted the original 2013 study and published a revised analysis that addressed the sampling errors that had been identified (3). As such, the source of error here is methodological (i.e., research design was complex; this increased the number of protocol items, processes, and likelihood of human error).
Estruch et al and PREDIMED conducted a review of how participants were assigned to various interventions groups. This effort revealed irregularities in their randomization procedures. As honest and responsible scientists, they withdrew the original report and published a new one. It is worth noting that once corrected the protocol deviations did not result in any significant change in estimates and inferences from the study, consistent with what one would expect from small random error from a very large study. To date, the original, retracted study has been cited close to 6000 times whereas the corrected one has been cited about 2000 times.
I read the original study when it was published in 2013 and its only because of this weeks assignment that I’ve learned about the retraction — pure chance!!!
PART B
As described by the authors in their original study, the PREDIMED trial was a parallel-group, multcenter, randomized trial (2). 7447 participants from Europe, Central or South America, and other countries across the world were randomly assigned in a 1:1:1 ratio to one of three dietary intervention groups across.
Was the randomization process performed at the individual level (i.e., 7447 independently sampled persons)? No, since the study included members from the same households, 11 study sites (i.e., intra-site correlations in statistical analysis), and in general inconsistent use of randomization procedures at some sites (4).
The revised analyses corrected the statistical correlations within families and clinics, omitted 1588 participants whose trial-group assignments were unknown or suspected to have departed from protocol, and estimates and p-values recalculated.
Despite these revelations no significant change in the results was observed. As mentioned earlier, this is somewhat consistent with the nature of errors arising randomly as contrasted with those arising systematically — as discussed in part A. That said, the discovery of these random errors highlights a self-correcting aspect already intrinsic to science, namely the systematic review and meta-analysis of already published works.
No recommendations will change the activity of the kind of welcome vigilantism demonstrated by Carlisle. One sees that its quite remarkable that Carlisle approached the issue from a purely statistical perspective and had an impact at the very highest tiers of pier review, the New England Journal of Medicine, and in a field of expertise (nutrition) that they may not have had any particular interest in! One also sees how responsive and responsible the authors Estruch et al were. And so this entire retraction story is quite reassuring in terms of the prospects of science or its current status.
However, I wish to recommend one other consideration. That we restore the old practice of reporting p-values in Table 1 of randomized trials, to demonstrate balance across trial arms. It used to be a standard practice across journals until p-values became unfashionable in certain high circles.
Also, I recommend that we restore the practice of using a flow chart to describe the sampling design of study participants. Such a chart as Figure 1 may have highlighted to reviewers and readers the sampling error committed by the authors and this might have been addressed in the peer-review period or in an editorial as early as 2013.
References
1.Carlisle JB. Data fabrication and other reasons for non-random sampling in 5078 randomized, controlled trials in anesthetic and general medical journals.
2.Estruch R et al. and PREDIMED Study Investigators. Primary prevention of cardiovascular disease with a Mediterranean diet. N English J Med. 2013 Apr 4;368(14):1279-90
3.Estruch R et al. and PREDIMED Study Investigators. Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virigin olive oil or nuts. N English J Med. 2018 Jun 21;378(25):e34
4.The Nutrition Source. Harvard T.H. Chan School of Public Health. https://www.hsph.harvard.edu/nutritionsource/2018/06/22/predimed-retraction-republication/Health AccessedNovember 9, 2022.
```
.
The odds tell me its due to chance, the gods are silent on this one!
11/09/22
One observed vs. expected mid-term election results for Senate and Congress
.
```
McArthur
11/11/22
We are gathered here, representatives of the major warring powers, to conclude a solemn agreement whereby peace may be restored.
The issues involving divergent ideals and ideologies have been determined on the battlefields of the world, and hence are not for our discussion or debate.
Nor is it for us here to meet, representing as we do a majority of the peoples of the earth, in a spirit of distrust, malice, or hatred.
But rather it is for us, both victors and vanquished, to rise to that higher dignity which alone befits the sacred purposes we are about to serve, committing all of our peoples unreservedly to faithful compliance with the undertakings they are here formally to assume.It is my earnest hope, and indeed the hope of all mankind, that from this solemn occasion a better world shall emerge out of the blood and carnage of the past -- a world founded upon faith and understanding, a world dedicated to the dignity of man and the fulfillment of his most cherished wish for freedom, tolerance, and justice.The terms and conditions upon which surrender of the Japanese Imperial Forces is here to be given and accepted are contained in the Instrument of Surrender now before you.
As Supreme Commander for the Allied Powers, I announce it my firm purpose, in the tradition of the countries I represent, to proceed in the discharge of my responsibilities with justice and tolerance, while taking all necessary dispositions to insure that the terms of surrender are fully, promptly, and faithfully complied with.
I now invite the representatives of the Emperor of Japan and the Japanese government and the Japanese Imperial General Headquarters to sign the Instrument of Surrender at the places indicated.
Let us pray that peace be now restored to the world, and that God will preserve it always.
These proceedings are closed.
```
.
```
11/12/22
Apropos of nothing
Being your slave, what should I do but tendUpon the hours and times of your desire?I have no precious time at all to spend,Nor services to do, till you require.Nor dare I chide the world-without-end hourWhilst I, my sovereign, watch the clock for you,Nor think the bitterness of absence sourWhen you have bid your servant once adieu;Nor dare I question with my jealous thoughtWhere you may be, or your affairs suppose,But, like a sad slave, stay and think of noughtSave, where you are how happy you make those.
So true a fool is love that in your will,Though you do any thing, he thinks no ill
— Woody of Louis
```
.
TV today
https://www.theguardian.com/tv-and-radio/2022/nov/14/john-oliver-british-monarchy
https://www.vulture.com/2020/04/woody-allen-apropos-of-nothing-review.html
.
RPPR
11/14/22
Login.gov (Google authenticator) .
eRA commons
Personal profile
RPPR… Menu
Route to next reviewer
.
Agism
11/14/22
https://www.vulture.com/2020/04/woody-allen-apropos-of-nothing-review.html
.
```
Hon. GAD K. GASAATURA Presents This Paper For Discussion:
In the recent past, some people, led by one Mr. Frank Gashumba, proposed that Banyarwanda of Uganda, as enshrined in the Constitution, be renamed 'Abavandimwe', (meaning 'brethren', of one womb/ mother). Such a move, they aver, will put an end to the discrimination, harassment and victimisation that Banyarwanda continue to be subjected to.
This presumes that the cause of discrimination is due to the obvious connotation in the word 'Banyarwanda', which might seem to connect them to Rwanda.
The proposal met with much bewilderment and consternation especially among Banyarwanda - both within and without the confines of Uganda..! It was widely rejected.
This article seeks to examine the issues raised in the said proposal.
Team Gashumba presents TWO ISSUES. And I will examine them within Two perspectives - The African Context & The Global Perspective:
The Main Issues Raised:
i) Many Banyarwanda of Uganda, are frustrated. Almost at every turn, they suffer much discrimination, and are treated as not being fully Ugandan; they often have to explain themselves as to why, if they are Banyarwanda, they do not want to go 'back home' - to Rwanda!
Consequently, many complain of being denied Uganda Passports, jobs, promotions and other services and opportunities ordinarily available to other citizens.
==========
As I will argue,
FEW BANYARWANDA WOULD DISAGREE WITH THE COMPLAINT.
==========
ii) In view of the above, Team Gashumba proposes that Banyarwanda of Uganda be Re-named 'Abavandimwe', to avoid being mistaken for Banyarwanda of Rwanda (Rwandans).
==========
While most will agree with the problem so identified:
FEW BANYARWANDA WOULD AGREE WITH THE PROPOSAL.. - to change their identity..!
==========
I will also introduce two perspectives:
iii) The AFRICAN CONTEXT - The Legacy of Colonial Boundaries:
iv) The GLOBAL PERSPECTIVE On Ethnic Rivalry & Conflict - Are We Alone..?
===================
TEAM GASHUMBA CONCERNS:
i) Are there Native Banyarwanda citizens of Uganda?
Refer to Population Census records in Uganda, starting in 1911; then 1921, 1931; 1948, then 1959 - before the influx of Refugees fro Rwanda, and 1969.
In all these Population Counts, Banyarwanda consistently turned out between 6% and 7% of the population, making it 6th or 7th largest ethnic group in Uganda, most of them in Buganda, Kigezi and Ankole.
Needless to say, There are, indeed, BANYARWANDA of UGANDA, equal citizens with the rest, who need not explain themselves more than is reasonable or as required of others.
As at 1893, there was no entity called Uganda. In present day western Uganda, there were several disparate Chiefdoms and Kingdoms, such as Mpororo, Nkore, Tooro & Bunyoro, which, in peace or in war, had close relations with other neighbouring kingdoms of Rwanda, Karagwe, Kooki, Buganda among others. Consequently, their respective people's would move across borders, to flee conflict or in search of opportunities.
In 1894, the British procured the first consent for 'British Protection' from Buganda. Other territories were subsequently added - Ankole, Tooro, Bunyoro etc. to form the 'Uganda British Protectorate' - until Independence in 1962.
The very first Constitution - The 1962 (Lancaster negotiated) Independence Constitution of Uganda established the 'Persons to become citizens of Uganda, on 9th October 1962.
It covers most Banyarwanda in Uganda.
============
1962: UGANDA INDEPENDENCE CONSTITUTION:
CHAPTER II: CITIZENSHIP 7(1):
"Every person who, having been born in Uganda, is on 8th October 1962 a citizen of the United Kingdom and Colonies or a British protected person, shall become a citizen of Uganda on 9th October 1962:
- "Provided that person shall not become a citizen of Uganda by virtue of this subsection, if neither of his parents was born in Uganda."
1995: UGANDA CONSTITUTION:
CHAPTER 3: CITIZENSHIP 9.
Citizens of Uganda:
"Every person who, on the commencement of this Constitution, is a citizen of Uganda shall continue to be such a citizen.
The several categories of Banyarwanda in Uganda have been articulated elsewhere - including:
- Those who were 'bordered/ fenced-in' by the new Colonial Borders of Uganda;
- The various migrations, before and after 1926 up to 1959;
- Refugees
- Other Migrant workers
If I may speak for most of my compatriots originally from areas bordering Rwanda -
- The Greater Kigezi and Greater Ankole, many of our fathers, grandfathers or close relatives were enlisted and served under the King's African Rifles (KAR); Others served under the Kingdom Offices, Others were good Tax payers etc. Our parents, grandparents and those among us who were alive, were indeed and truly "British Protected Persons", on 8th October, 1962, who became citizens on the 9th October 1962.
My own maternal grandfather, AZARIYA KANYAMIBWA, was present, at the Kamwezi signing of the Protocol between Great Britain and Germany, Describing the Frontier between the Uganda Protectorate and German East Africa, Signed at Kamwezi in 1911.
He was also one of the Local Officials who accompanied the Enganzi (Primier) of Ankole, NUWA MBAGUTA, together with Europeans, in the demarcation of the Uganda/ Rwanda border. But my family was not alone in the border area; until 1960s, Banyarwanda made up a majority along the Rwanda border.
Notwithstanding the obvious explanations, however, reports and complaints abound, of harassment and discrimination that Banyarwanda in Uganda continue to suffer, especially when they need to access public services. It is wrong, to regularly require greater proof of citizenship, for instance, from members of one particular ethnic community than is asked of others.
ii) SHOULD BANYARWANDA of UGANDA CHANGE NAME?
.
As has been stated elsewhere by many Banyarwanda, the suggestion is not only uninformed, it is unreasonable and ridiculous:
- It is uninformed, because most of the offenders do it out of malice; It would only be fair to expect them still to continue in their 'ill will' machinations against the Banyarwanda community, no matter the name.
Many still recall the 'Ethnic Cleansing' of 1982, in which more than 100,000 Banyarwanda were expelled by the UPC Government and UPC functionaries, especially from 'Greater Ankole..!' Citizens were forced into Refugee Camps in Uganda, while others were driven across the border into Rwanda - only to die in thousands, in poorly managed refugee camps.
Among those expelled, were thousands who had completely assimilated into other communities, who could no longer speak or comprehend The Kinyarwanda language. To many who witnessed the treachery of neighbour against neighbour, in Communities and would-be Places of Refuge, the Churches, the 'Name Change' suggestion just comes as a 'Bad Joke..!'
Yes, the suggestion is ridiculous, because the Banyarwanda is a nation of people, spread across the East and Central African states, and around the world; they are native in the D.R.Congo, in Rwanda, Tanzania and Uganda; Kinyarwanda/ Kirundi is probably third most widely spoken language in Africa - after Arabic and Swahili; sizable populations of Banyarwanda has also grown in other parts of Africa, Europe and the Americas.
Banyarwanda are a Nation of people, scattered by circumstances throughout the world, but whose Culture is a great source of Strength to be sought - not to be shunned..!! There're many people groups that would 'kill to be allowed into' such a MILLENIUM-OLD kith and kin association.
Just to demonstrate one element of this Community Strength that many members will already be familiar with - "GUSHUMBUSH' INKA":
The Banyarwanda Culture compels community members 'Kugir' UBUNTU n' UBUPFURA..!!'
(To be kind-hearted and considerate of others);
Custom requires one to be mindful of one's neighbour's dire needs; your neighbour is to be treated as your own brother, whose nakedness, hunger or want becomes your own nakedness and shame..! Others will think you less, if you do not reach out..! So powerful and compelling..!
If, for instance, your neighbour or relative fell on hard times - lost most of his (cattle) wealth, his fault or not, it becomes your duty to work it out with him and others, as to how best to quickly cover his shame and need - to keep that man's, and community dignity.
Banyarwanda would give him cows - to be returned after 3 to 7 years or much longer, when the man has been able to get back on his 'feet' - "KWITURA".
This, to the envy of many.., protesting:
.... "KWONK' ABANYARWANDA NIMUKUNDANA..!
..... "NAYE KALE, ABANYALWANDA MWAGALANA..!
Is this a culture and name to abandon and to shun?
I say, NO..!
TUR' ABANYARWANDA;
TUR' ABAVANDIMWE;
TUR' IMFURA Z'ABADAHEMUKA..!!
iii) THE AFRICA CONTEXT - The Legacy of Arbitrary Colonial Boundaries:
On all borders of Uganda, as oft the case for most African countries, one finds communities that were separated by The Arbitrary 'Pen of The 1884/85 Berlin Conference', wherein the European Powers greedily partitioned and distributed the African lands among themselves - like hyenas on the weakened buffalo..!
Already, Communities were also often found straddled across recognised frontiers of pre-colonial kingdoms. People would move to flee conflict or in search of food and pasture.
With the Partition of Africa - Uganda, like most other African colonies, was handed artificial border lines that literally split families and communities apart..!
In Kenya, they had to contend with the question of 'KENYAN SOMALI' natives of Northern Kenya. In Angola, they have a large native population of 'BAKONGO' - same people as those in Congo (DRC) and in the other Congo Republic; In South Africa, there's a native 'SOTHO Tribe' - kinsmen to the people of LESOTHO. SO WE ARE NOT ALONE..!
Similarly in UGANDA: We have, at our borders, the same populations as the neighbouring countries. Kenya and Uganda share several ethnic communities: Samia, Bakusu/ Bagisu, Iteso etc.
There was an interesting story in The Daily Nation of Kenya recently:
"A Tale of Two UGANDA Kings Who Hold Big Sway In Kenya Politics:
Cultural kings are recognised in the Ugandan Constitution — not in Kenya — but traditional powers bestowed upon two kings in Eastern Uganda spill over to Kenya.
One King, of the Bamasaba brings together the Bagisu of Uganda and Bukusu of Kenya of a common ancestry, King Wamimbi; and the King of the Itesos of Kenya and Uganda, His Highness Papa Emormor; Both have commanding respect among their subjects inside Uganda and Kenya.
In the last Kenya elections, a Ugandan cultural king was implored to intervene and strike a truce between two senior political leaders who were at loggerheads in Western Kenya - Ministers Moses Wetang’ula (Trade) and Eugene Wamalwa (Justice and Constitutional Affairs), both from the Bukusu community, were key pillars for Cord and Jubilee coalition campaigns in the region.
“I was approached by some leaders from Kenya to reconcile the two politicians..,” Bamasaba King Wilson Wamimbi (Omukuka) told The Standard on Sunday in an interview at his Busano home near Mbale town, Eastern Uganda.
But perhaps the most dramatic case to demonstrate the travesty served the African communities, is that of the renowned Awori family - also on the Kenya border.
The Tale of two Awori Brothers:
MOODY AWORI (b. 1928-) was the 9th Vice President of Kenya under President Kibaki; And
AGGREY AWORI (b. 1939) former MP, Minister for ICT & former Presidential Candidate in Uganda..!
The two brothers, from a family and clan that straddles across what was later to become the Uganda-Kenya border are sons of Rev. Canon Jeremiah Musungu Awori, a pioneer priest of what was then known as the Native Anglican Church (NAC) in East Africa, and his wife,
Mrs. Mariamu Odongo Awori, a nurse and community teacher. Rev. Canon Jeremiah Musungu Awori was son of Awori Khatamonga - a fabled elephant hunter who operated at the Uganda-Kenya border from the mid-1800s.
At one time, the two brothers - Aggrey Awori & Moody Awori were serving respectively in Uganda Cabinet and Kenya Cabinet; One was running for President in Uganda, the other became Kenya's Vice President.
There're many other similar cases, albeit less dramatic:
A classmate of mine at King's College, Budo, from Sebei, like many Ugandans, fled to Kenya, during Gen. Idi Amin's mayhem. On reaching Nairobi, he became a Kalenjin - President Moi's ethnic group..! Another friend of mine, from Karamoja, on completing studies in Mbale and Makerere University, went to Kenya and quickly became a Pokot - he's still serving in Cabinet..!
Our own Hon. Dr. Chebrot, my classmate and former State Minister, was very much at home in Kenya, until opportunities improved in Uganda.
The same can be said of almost every borderline: The ITESO people, for instance, are to be found in 'Greater Teso' of Uganda; But there is also a large community of ITESO people who inhabit Tororo and Busia areas; And across the border, in Kenya, we find a district called 'ITESO' District mostly populated by the Iteso..!
Similarly: A large population of the ACHOLI people in South Sudan; the LUGBARA, ALUR, BATOORO/ BAHUMA & several others, are also native in the Democratic Republic of Congo..!!
In Europe: Switzerland has four tribes: Germans, French and Italians and the Romanche;
Belgium has a large French tribe; Austria and Hungary, the Germans.
On Uganda's Southern Flank: The people of KARAGWE in Tanzania, are quite similar to the people of ANKOLE & KOOKI (RAKAI District).
Still on south western border of Uganda, is RWANDA, with whom Uganda has had much interaction for centuries - in war and in peace. Consequently, many Runyankore/ Rukiga speaking communities were caught on the Rwanda side; and many Kinyarwanda speaking people were 'Demarcated' inside Uganda, in addition to others, in Ntungamo, Rukiga, Kabale & Kisoro, whose territories were adjusted and place inside Uganda, by European Agreements.
Former Rwanda President HABYARIMANA, for example, was a MUKIGA from Gisenyi, North-western Rwanda, bordering Kigezi.
To the North-East of Rwanda, is the Umutara province, where native Runyankore speaking cattle keeping (Bahima) of the Bagina, Bashambo and other clans were settled. Some of them - Kataraiha and Kisiribombo - were appointed by the Rwanda King as Superior Chiefs. Most of these Bahima communities remained in Rwanda until the 1990/ '94 war.
iv) THE GLOBAL PERSPECTIVE ON ETHNIC RIVALRY And CONFLICT:
From the end of WWII in 1945 to the 'Fall of The Berlin Wall' in 1989, Global Conflict was mostly driven by 'The Cold War' between The USA and USSR.
But now all over the world, especially in the last 30 years, the world is at war with itself - Conflict mostly driven by Nationalist Extremism. Ethnic conflict has been cause of much suffering, war & destruction.
Nationalism was principally responsible for starting both WWI & WWII.
By 1900, the Austria-Hungary Empire was the largest and most powerful political entity in mainland Europe, holding together a dozen disparate ethnic communities, including: Germans, Hungarians, Polish, Czech, Ukrainians, Slovak, Slovene, Croatians, Serbs, Italians and the Romanians.
The Slovacs nationalists in Bosnia and Herzegovina, however, were no longer happy under the Austria-Hungary rule; they considered themselves Serbians, and preferred to have their territory ruled by their kith, in Serbia.
Over time, tensions rose. A Slovac terrorist assassinated an Austria-Hungarian Duke. Then war was launched: Austria declared war on Serbia, which quickly drew in Germany, Russia, Britain, France .. The rest is just history, of 10 million dead, 25 million maimed. And a Global Economy on its knees. ALL FOR TRIBAL/ Ethnic ANIMOSITIES...!!
Banyarwanda of Uganda are not alone..!!
Many countries seem to be struggling with nationalist sentiments. The world headlines are filled with Nationalist Conflicts, laden with Ethnic emotions:
Whether you look at Germany or France; the Uighur Muslims in China, the Political contests in South Sudan, the Rohingya Muslims of Myanmar. The world needs a considered response.
In the United Staes of America, 'Black Lives Matter' is the Battle cry for African-Americans; and now Asian-Americans seem to be the immediate target. The world is aflame with tribal rage.
Personally, I think one of the most backward element included in our Constitution was the promotion of Ethnic loyalties - requiring even the most patriotic Ugandan to first identify with some tribal grouping..!!
A most unfortunate inclusion..!
=======================
ADDED:
Excerpts From Counsel EDGAR TABARO, son of Retired Justice Tabaro:
".. Early recorded migrations of Banyarwanda to Uganda in their hordes was at the beginning of the first decade of the 20th Century, when cash crop was introduced by Mitchel Cotts the successor to the Uganda Company (incorporated 1896) itself a successor to the Imperial British East Africa Company (IBEACO) that had a royal charter. The early migrants are mainly in areas of tea production in Namutamba and Tooro where they worked as wage earners although a good number took up other menial jobs in other counties of Buganda and their descendant continue to live.
In 1910, the Anglo - German – Belgian conference was held in Brussels, which resulted in several agreements ... between Belgium and Germany .. One, signed at Brussels, on May 14, 1910, settling the boundary between German East Africa and the Belgian Colony of the Congo.
Another Signed at Brussels, August 11, 1910, further on a Protocol between Great Britain and Germany Describing the Frontier between the Uganda Protectorate and German East Africa. Finally confirmed, Signed at Kamwezi, October 30, 1911.
Memorandum attached to the Protocol List of Boundary Pillars on the Anglo - German Boundary, Sabinio to River Chizingo, with approximate Co-ordinates.
These two legal documents transferred territories of Rwanda Kingdom Provinces of Bufumbira (present day Kisoro District), and Ndorwa (present day Kabale, Rubanda and Rukiga Districts), and areas of Ntungamo that were inhabited by native Banyarwanda communities.
It is worth noting that these are the persons referred to as Banyarwanda, an the indigenous community of Uganda as per the 3rd Schedule to the Constitution, though the ones of Bufumbira (later) assumed the identity of Bafumbira.
For a long time under both colonial and post-colonial administrations in Uganda, the areas inhabited by the Banyarwanda communities did not benefit from any form of affirmative action, a result of which the community was heavily impoverished.
Infact, it was a deliberate government policy to reserve the areas as a cheap source for labour for the plantations and other richer agricultural regions. However, over the years, descendants of these people, (through hard work and discipline) have been able to lift themselves out of the shackles of poverty and ignorance, which may not necessarily endear them to other communities, hence exacerbating their resentment.
To this end, the Constitution of the Republic of Uganda, the following persons shall be citizens of Uganda by birth—
(a) Every person born in Uganda one of whose parents or grandparent/s is or was a member of any of the indigenous communities existing and residing within the borders of Ugandaas at the first day of February, 1926, and set out in the Third Schedule to this Constitution.
Uganda’s indigenous communities as at February, 1 1926 which forms the date to the last border adjustment to the Protectorate of Uganda that transferred the Eastern Province to the Kenya Colony, which is now the Rift Valley up to Lake Rudolf (Turkana).
This 1926 adjustment is at the centre of the conflict over Migingo Islands!
```
.
Agism
11/15/22
Like many of our fathers and grandfathers, Allen is a 20th-century man in a 21st-century world. His friends should have warned him that “Apropos of Nothing” is incredibly, unbelievably tone deaf on the subject of women
.
Jon Stewart
11/16/22
A white persons success is because of privilege
A minorities success is empowerment
A Jews success, that’s a conspiracy!
.
```
What needs to happen to foster positive change in regard to research climate, culture and conduct in science, for intellectual honesty and integrity on all levels of scientific practice, as well as in society, to become the default for all undertakings?
I think the philosophy underpinning statistics must be addressed to foster positive change in regard to intellectual honesty and integrity of science.
Two schools of philosophy exist in the world of statistical inference: Frequentist and Bayesian (1).
To those unfamiliar with these schools I’m 100% certain that your exposure to statistics has been Frequentist. And to those familiar with these schools, it is without a doubt to any of you that the chief reason Bayesian statistics are not routinely taught at most levels of education (including most doctoral programs at as prestigious an institution of Johns Hopkins) is because they are complex (2).
But what is universally understood about these two schools of statistical inference? Fundamentally, the frequentist school relies too heavily on any single study for statistical inference whereas the Bayesian school formalizes our prior beliefs, then formalizes a statistical interaction between the strength of those prior beliefs and the latest findings from the latest study, and then the prior beliefs are thereby updated.
Sounds complex? Yeah, that’s why they don’t teach these methods! And therein lies the problem (3). As a community, we have chosen the easy path of Frequentist statistics that relies exclusively on one study, arbitrary p-values, and all the machinations of the authors of that study including their rigor, their proneness to error, their sloppiness, and their integrity. The tough way of Bayesian statistics is always a community effort, codified in the Bayesian prior knowledge, which always tempers the new information from the new study.
A fraudster who understands Bayesian statistics and who also is aware that peer reviewers and the audience at large are all masters of Bayesian statistics will be preempted from committing fraud. This would mean that teaching Bayesian statistics per se would be a preemptive solution to fraud rather than a reactive one, something described by the Director of the Office of Science Quality and Integrity as the ideal solution (4).
On a conservative note, Frequentists have met Bayesian halfway using meta-analysis to pool inferences from several studies and to reach one unifying conclusion about our best estimate of an effect size and the degree of our uncertainty (or heterogeneity across studies). Perhaps if these frequentist methods were not restricted to advanced courses, then fraudsters may also realize that their individual efforts will be fraught if they differ substantively from the rest of the literature. In other words, a fraudster would be certain to be found out!
To conclude, I am pessimistic about the moral integrity of the individual scientist for reasons outlined in my previous two posts. But I’m optimistic about the collective integrity of science for reasons also outlined in my previous post, which today I formally christen “Bayesian inference.”
References
1.Harrington D, et al. New Guidelines for Statistical Reporting in the Journal. N ENGL J MED 381;3. July 18, 2019
2.Brophy J. Bayes meets the NEJM. https://www.brophyj.com/files/med_lecture_bayes.pdf Accessed on November 16, 2022
3.Rosenquist JN. The Stress of Bayesian Medicine — Uncomfortable Uncertainty in the Face of Covid-19. N ENGL J MED 384;1 January 7, 2021
4.Thornill A. What’s all the Fuss About Scientific Integrity? https://www.doi.gov/ppa/seminar_series/video/whats-all-the-fuss-about-scientific-integrity Accessed on November 16, 2022
```
.
Dirty Harry
Quintessential Don Siegel protagonist
14/26
Law enforcement (Antagonist) vs Criminal (Protagonist)
Written in favor of the scum
Society crying police brutality
Public is siding with the crooks
30-32/10
Black militants
Lead protagonists you find it difficult to root for
It’s takes a magnificent film maker
To thoroughly corrupt an audience
.
Emma Goldman
11/17/22
But I insist that not the handful of parasites, but the mass itself is responsible for this horrible state of affairs. It clings to its masters, loves the whip, and is the first to cry Crucify!
— Emma Goldman
Ref.
When you go to woman, take the whip along
— Nietzsche
.
```
There are two outlooks to life. One is of the ascetic priest and involves self-denial of the innate pleasures derived from food, sex, and violence. The other is cut-throat and is marked by expenditure of energy on a worthy enemy, recuperated by way of the spoils of war, and an unquenchable lust for the next battle.
One outlook is more likely to yield modern day “struggles” against mental health, obesity, cardiovascular disease, and cancer. The ascetic priest might win their several “battles” against our species-essence and achieve longevity following a consistent spiritual and physical life-course marked by self-denial. But their followers will often fall short of expectations in this metaphysical battle; thereby becoming anxious, guilty, and alas, both spiritually and physically ill.
More inline with our innate predilections, the warriors outlook can be represented as the military school of life: that which doesn’t kill you makes you stronger. So the option any person or tribe or nation faces is simple: keep fighting.. or yield (to unnatural asceticism or to the enemy).
Between these two stark options one will find the marvelous delusions that civilized man has crafted. Pacifism, United Nations, World Trade Organization, War Crimes Tribunals, et cetera. We might then conclude that modern man is at risk of completely alienating himself from his essence, were it not for creative solutions that rescue him from despair — such as the Olympics, the World Cup, Boxing 🥊, the cult of exercise, or the depiction of violence in film and literature. The cult of Hero worship is the vestige of a species that coulda woulda shoulda been warrior-like, were it not for its frailty. Oh frailty, thy name is man!
And so one may ask whether mans’ creative excuses achieve their cathartic purpose. Or whether they merely postpone the inevitable regression to our species essence, while making man more frail, diseased, and ill-suited for this purpose. But such are the questions one needs to ask to restore vitality and meaning to life. Only then might the taunting & mocking of childhood make sense, and only then might the bullying and humiliating initiations rites in teenage & fraternities find a context, and of course only then might the fierce competition amongst high school girls & just as much between corporate be understood as a mere side-show — to the original cut-throat essence of the species. Participants in these “battles amongst lost sheep” most certainly will seek a priest or therapist or opium dealer at some point in their life. Without expending a sufficient amount of energy on overpowering a worthy enemy, the modern man hoards his energy and — because E=mc2 — locks it in as obesity. All the prescriptions of the ascetic priest (really perpetuating factors of disease if you think of it) — purging oneself of food, the moralization of sex, and the proposition of kindness as the highest virtue — glorify hibernation. Increased life expectancy thus becomes the proof of progress. And thats how a life is put to complete waste.. in the pursuit of some ideal where there is no blood.
```
.
What can I say?
The dude abides!
http://dudespaper.com/because-whats-a-hero-in-defense-of-slackers.html/
Famous slackers:
1. Lao Tzu
2. Jesus the Nazarene
3. Diogenes
4. Buddha
5. The Dude
.
Advertised as the “slowest-growing religion in the world”, Dudeism offers the chance for anyone to be ordained for free on their website.
.
```
META SCIENCE
Using research methods to study science itself
11/30/22
Last week I suggested that we apply a scientific method of inquiry into the Open Science Recommendations so that we do not take our dear reproducibility activists at their own word.
I had no clue that I was already dabbling in meta science. So it really is possible to actively engage in an endeavor without having a name or definition for it. And so the 93 minutes of the first Youtube video dedicated to the question “What is Metascience?” tells me that perhaps our time and effort here could have been spent on more practical issues.
The crux of the 93 minute video is that metascience is research on research. That for me is enough. The object of interest is scientific research. And the investigators who do research on research are doing meta science. What methods do they employ? What is the epistemology? They should be in essence the same used in science including trials, experiments, statistical analyses of findings, inferences, and conclusions.
One discussant has a vision of meta science as being interdisciplinary, with no boundaries across science fields. While I will not discuss this in detail here, I am not at all optimistic of this proposal because in the real world funding and financing of science is key to its sustenance. And the proposed cross-disciplinary meta science is likely to remain an orphan since each science is sui generis — has its own unique set of issues, challenges, and most pressing questions — none of which is meta science.
What research topic would I choose to address if I had unlimited means? I would choose to develop an automated methodology that generates a meta-analysis of each paper submitted for peer-review to tell us how the inferences from the meta-analysis look before and after the new study is incorporated into the scientific literature. I’d then ask the authors, reviewers, and journal editors to write a one-sentence speculation on what makes this study unique to the existing literature.
Anyone familiar with Lancet, JAMA, and several top-tier medical journals knows that they already publish a brief research-in-context panel (What was known before? What does this paper contribute? And what remains unknown?) (1). What I propose is quantitive and reported with a classic forest-plot
My proposition would be embedded within existing research frameworks, modes of publication, and methods, and would only face opposition if it entailed extra effort for researchers. Hence, I envision this to be an automated process. The authors of the original science or the journal editors would only need to enter key items such as study outcome, risk factors, and outcome metrics. This would be original research but also placed in a broader context as research on research (i.e., meta-analysis and commentary).
References
1.Information for Author Item 9. Lancet. https://www.thelancet.com/pb/assets/raw/Lancet/authors/tl-info-for-authors-1660039239213.pdf. Accessed November 30, 2022.
```
.
Anatomy
11/30/22
1. Truth
2. Rigor
3. Error
4. Sloppy
5. Fraud
.
No reason to be confused
12/02/22
1. Dostoevsky
2. Oscar Wilde
3. Roman Polanski
4. Woody Allen
5. Michael Jackson
6. Kelly (I’m flirt, Age ain’t a thing)
To mention but a few of the greatest artists of my lifetime.
Great art is at its best seduction, corruption of a naive world view
And the artist will run afoul of the law in autocracies and democracies alike
Shakespeare is the supreme artist whose wide-range of characters were always thought of as distinct from him (no single man can be all those kings, low life’s, women, teenagers, solders, lovers at once)
Perhaps his life remains a mystery for the good of literature. For what mores may come forth when you set day light upon his magic?[^35]
[^35]: man $\ne$ artist
.
#### 12/2022
```
Comedy vs. Tragedy
12/01/22
Whence evil?
The answer!
United these :)
Views:
1.Tragic — everything is meaningless. So there’s no such thing as evil. Its just bad luck
2.Comic — mistaken identity. Most recluse fella isn’t immune to mistaken identity
3.Evil — in other words, evil mistakes the recluse for some other it is targeting
— Midsummer nights dream
—
```
.
[ambitious](https://en.wikipedia.org/wiki/John_Singleton#Early_life): comic books, video games and movies
q&a types: vs (a potential buffer against)
slackers: drugs, the partying and shit
.
Saidi
12/02/22
13.1 miles
103 minutes
7’51” per mile
.
Boyz N the Hood
“One out of every twenty-one Black American males will be murdered in their lifetime”
“Most will die at the hands of another Black male.”
Opening sequence in John Singleton’s “Boyz N the Hood” — 1991
Duncan?
.
Woody Allen’s 10 justifications for living (Manhattan)
12/03/22
Might we add [Jimmy Fallon](https://www.youtube.com/watch?v=ziv8CYsbYWU)?
.
Selena Gomez
12/06/22
1. Bipolar
2. Lupus
3. Kidney failure
4. Transplantation
5. Celebrity
6. Millions in the bank
.
Once upon a time in Hollywood (The Book)
12/06/22
You can call me Mirabella
Roman Polanski
Sexy evil hamlet
How can Mirabella be Ophelia???
Trudy Frazer on Rick Dalton’s lap
Narrator treats 12yo as mature
One day 8yo trudy looked her age (i.e. young)
Undertones of Nabokov are evident
It’s takes a magnificent film maker
To thoroughly corrupt an audience
— Dirty Harry, Cinema Speculation
.
Hilton
152 West 26th St
.
Tarantino’s work appeals to resentment
No wonder historical fantasy is a key part of it
And commercial success is something his work often enjoys
.
Auteurs
12/06/22
Pessimistic/Absurd: Coen Brothers, Iñárritu
Resentment/Historical Fantasy: Tarantino
Slacker/Epicurean: Woody Allen
.
```
Elements of ethical science communication
12/07/22
Part I:
I think a fundamental logical fallacy in “Big Sugar’s Secret Ally? Nutritionists” is to mistake cause with effect (1). The basic hypothesis of the article is that the 655% increase in prevalence of Americans with diabetes over the last 60 years is caused by an increase in consumption of sugar.
However, the true causal relationship is that diabetes causes an increase in blood sugar levels, not the other way around wherein increased blood sugar levels (from dietary sources) increase likelihood of diabetes.
What causes diabetes? More specifically, what causes type-II diabetes (the epidemic kind)? The answer is an increased resistance to the effects of insulin, arising often from a change in body composition with more percent body fat than muscle. Adipose tissue is insulin resistant.
And the cause of increased body fat over the last 60 years is reduced physical activity and more or less sedentary life style amongst Americans of most ages.
To say obesity is caused “by a lack of energy balance” is incontestable. The article correctly quotes the NIH in saying “in other words, by our taking in more calories than we expend.” But then the author reveals their bias by focusing on the “taking in more calories” and completely neglecting the “more than we expend.” Hence, the entire article focuses on how food may influence our body weight through their caloric content.”
But how about how increased activity and exercise may moderate our appetite and thus our calorie intake (2)? This question uncovers the authors biased question and article title. It blinds us to the cause of dietary intake, namely, the hormones and endorphins that regulate our appetite in the first place.
Modern persons at all the stages of life including childhood, teenage, young adulthood, middle life, and later life have drastically reduced outdoor activity. Many more more video games, increased indoor entertainment through television, sprawling of suburbs and reliance on cars for every mode of movement (from home to grocery store, to school, to work, etc.) and such pattern have increased exponentially over the last 60 years and are the fundamental problem. Sedentary people eat more than active ones. So dietary intake is an effect arising from physical inactivity.
Of course the consequence of the logical fallacy discussed here is the focus on diet and nutrition instead of physical activity, lifestyle, and environmental engineering that may nudge individuals back to what their ancestors did: more physical activity for daily tasks.
Part II:
My favorite example of a statistical fallacy comes from American politics but may as well apply to clinical studies. Over the last 40 years, the American senate has had either a majority democrat or republican representation. But by majority I mean 51% (majority party) vs. 49% (minority party) of the 100 senators, two from each state. Just today, with Georgia’s senate runoff election complete, the democrats hold 51 of the 100 senate seats (3).
Those is the majority interpret this as the electoral mandate of the people who have chosen them over their rivals.
But when you subject this to statistical inference this is essentially not different from the toss of a coin or a random electoral event bearing absolutely no meaning. When you toss a coin 100 times (to elect the senators), that may count as a sample size of n=100. And the majority of 51% may count as p=0.51. The standard error is given by the square root of p(1-p)/n, 0.05. Thus such a slim majority is 51% (95% CI: 41.2%-60.8%).
Here we see that this isn’t statistically significantly different from the null-hypothesis from a coin toss (pure chance, luck) of 50%.
References
1.Gary Taubes. Big Sugar’s Secret Ally? Nutritionists. The New York Times. Jan. 13, 2017
2.James Dorling, et al. Acute and chronic effects of exercise on appetite, energy intake, appetite-related hormones: the modulating effect of adiposity, sex, and habitual physical activity. Nutrients. 2018 Sp; 10(9): 1140
3.Georgia’s U.S. Senate Runoff Election Results. The New York Times. https://www.nytimes.com/interactive/2022/12/06/us/elections/results-georgia-us-senate-runoff.html. Accessed December 07, 2022.
```
.
Best swim ever
12/08/22
See lose.it
.
Mbappe 1/2 -200
Messi 7/2 +350
.
tweyanziza tweyanzege
.
Good [luck](https://www.ft.com/content/f5228f5e-11ab-11e6-91da-096d89bd2173) (England)
Bad luck (Brazil)
.
Tirzepatide for treatment of obesity NEJM
Semaglutide in adults with overweight or obesity
NEJM
.
That’s the church address
237 W 51 st, New York, NY 10019
.
Spontaneous [skill](https://www.cnn.com/2020/07/12/football/ronaldo-brazil-world-cup-final-1998-cmd-spt-intl/index.html) still stirs the soul for footballers everywhere
.
```
Censure
12/12/22
i guess i have a more playful approach to (not being but… )
becoming a better member of the audience:
my sport is to take what the performer mistakes
“Art being to a certain degree a mode of acting, an attempt to realise one's own personality on some imaginative plane out of reach of the trammelling accidents and limitations of real life, to censure an artist for a forgery was to confuse an ethical with an aesthetical problem.”
― Oscar Wilde, The Portrait of Mr. W.H.
Do we mistake an aesthetic issue
For an ethical issue?
Can we be offended by
A horrid performance?
This is perhaps my Q!
```
[^36]
[^36]: nothing succeeds if [prankishness](https://www.gutenberg.org/files/52263/52263-h/52263-h.htm) has no part in it
.
Ground rules
12/12/22
1. Everyone has good intensions
2. Confidentiality of meeting
3. If you must share then no names
4. Conversation, nodding, finger snap
5. How did it make you feel? (reactive)
.
1. Comedy & tragedy are the same
2. So maybe jokes in science be avoided
3. General casual demeanor is wrong
4. Let’s stick to O, E, M, C, S (epistemology)
5. May consider some surrogates, pending..
.
1. Dry detached scientific presentation
2. Very highly structured ABMRDB
3. Lack of boundaries when working?
4. Character, anecdotes, like liability of scientist?
5. Fully circle with pathos, logos, ethos
.
I think its ok for girls and women to talk feelings
But if you want to know guys feelings?
You may not like what you hear!
.
Instead of treating audience as passive with no say
Let’s separate dogmatists from don juans (proactive)
Treat all performances as a ruse (suppose truth a woman!)
.
Pessimism (this lone wolf) vs.
Optimism (most JHU-TRC)
Aesthetic (P) vs. Moral (O)
.
How did it make you feel?
12/12/22
Pessimist: I felt nothing since there was no aesthetic merit
Optimists: went against the TRCs core values and we felt uncomfortable
Censure: Art/life is a mode of acting, we confuse an ethical with an aesthetical
Henry IV Part I: “counterfeit” 13 x of 71 times in Shakespeare!!
That’s 18% of his usage in just one play (2.6% of oeuvre)
Which is 6.9-fold greater than by pure chance
.
bet365
Argentina @5/6, -120
France @5/4, +125
Morocco @11/1, +1100
Mbappe @4/5, -125
Messi @3/2, +150
Giroud @10/1, +1000
.
Aly Strauss
12/14/22
Start and/or end with human subjects
That’s what makes GTPCI/Data science different from epidemiology and microarrays and health services
Key word: interface (visual, apps, etc)
Machine learning/AI is now FDI approvable because its a medical device that impacts practice
Research involving a consent process is the bottom line minimum…. Not meeting definition of GTPCI
Provider consent, which is my calculator, and aim 3, is perhaps my Segway into HSR
Data science for public health and most of the class are engineers, can do just the first 4 of 8 credits - TERM 3
Causal inference TERM 3 (online version in Term 4)
.
Flu or COVID
12/15/22
1. Sore throat
2. Nasal congestion
3. Muscle pain
4. Fever
5. Malaise
6. Anosmia
.
Trump's major announcement is that he's finally ready to unveil his alternative to Obamacare - DonTcare.
Which is exactly how most of us feel about his announcement.
.
Two degrees of delegation
Samuel Benjamin Bankman-Fried
His aunt Linda P. Fried is the dean of Columbia University Mailman School of Public Health
.
Muzaale contra Aristotle
12/15/22
force, counterfeiting: one tameth chaotic nature by some contrivance
epistemology: must demonstrate passion, implies attention to detail
ontology: need consent since one hideth it in name of privacy
.
Wakayima vs Enkuddu
Obule embiro
Obbe namagezi
.
Agility
Intellect
.
Myself or a household member has tested positive for COVID-19 within the past 14 days. Should I report for jury duty?
DO NOT REPORT if you or a member of your household has tested positive for COVID-19, if you are experiencing symptoms, or if you are waiting for COVID-19 test results. Please call the jury office at 410-333-3775 to reschedule your service. The call center is to accept calls Monday through Friday, between 10:00 a.m. and 4:00 p.m., except State and Federal holidays.
https://www.baltimorecitycourt.org/wp-content/uploads/2022/04/Answers-to-Questions-Jurors-May-Ask-During-Reopening-03.30.22.pdf
.
Jury Duty
12/17/22
Called 410-333-3775
Voice note stated that
Reporting #s 6000-6999
Should report today
Mine is 8353
.
Interesting
12/17/22
This charged atmosphere has produced a backlash from two groups of people who rarely agree on anything: conservatives and famous comedians, who believe that sensitivity to offense is stifling free speech in comedy
.
idiom old-fashioned humorous. to play the piano: He's the piano teacher who taught my daughter to tickle the ivories.
.
Babyface
12/17/22
Man child
Crowd pleasers
Deele (leggings, makeup, soul Glo)
Learning ground
Crowd reactions
Jerry curl (harms hair)
California curl (looks natural)
The moment you feel fake
You’ll lose your audience
People will dismiss you
.
R Kelly & Lolita
12/17/22
https://www.vox.com/culture/22621692/aaliyah-death-20-year-anniversary-r-kelly-trial
https://medium.com/@mslola/surviving-lolita-adapting-nabokovs-pentapod-monsters-for-the-metoo-era-9d781a8dd1d3
https://rachel-wagner.com/2020/09/27/lolita-r-kelly-me/
.
Realedy mocks idleness
Falstaff exposes what is counterfeit in
The ambitious and the moral (Justice shallow)
Orson Wells
— Chimes at Midnight
Honor: thus ends my catechism :)
.
Hotspur , Tragedy
King Henry IV , Comedy
Falstaff , Realedy (Give me life!)
.
Slackers mock both
https://en.m.wikipedia.org/wiki/Chimes_at_Midnight
.
Relatable
12/19/22
[Navel gazers](https://www.emory.edu/EMORY_REPORT/erarchive/1999/April/erapril.5/4_5_99brown.html) unite!
```
Emory Report
April 5, 1999
Volume 51, No. 26
First person:
Reflections on an over-examined life: navel gazers unite!
It's high time someone gave us political recognition. Not that we'd ever actually vote, of course. We're far too busy working through The Artist's Way to leave the apartment for anything other than brie and cigarettes. But we like the idea of voting. We like the idea that social change is but an election ballot away--and personal transformation is as easy as e-mail.
If television personality Ally McBeal and radio therapist Dr. Laura Schlessinger were inverse halves of the same brain, they would exemplify the mind of the Postmodern Navel Gazer: the gourmet cheese-loving, expensively educated, Gen-X individualists who spend our waking hours trying to have our pain and ease it too. Heads without bodies (with sporadic alcohol-induced reversals), we applaud Dr. Laura's self-help sound bites while at the same time empathizing with Ally's endless neuroses. Is this fuzz in my navel lint or inconsistency?
At least three assumptions characterize the navel gazing mentality as I understand it:
Assumption 1: You believe that desire is better than satiation and take steps to ensure that you always want that which you can never have. Happiness is not sexy. Longing is.
Assumption 2: You boast a nihilistic exterior yet secretly believe The Truth is out there. But you want X-Files Agent Scully to find it for you. She's not a poet and she's not emaciated, so she has more time and energy than you to do that sort of thing.
Assumption 3: You gravitate toward quick-fix solutions to complex psychological and social problems. This Minute Rice approach helps ensure that while you continue to self-destruct, you can relax in the belief that change is just around the bend--should you ever decide to get out of bed and look for it.
Take my friend Gwen as an example. Gwen worships Dr. Laura with religious intensity. Her bookshelf displays Ten Stupid Things Women Do to Mess Up Their Lives at one end and, to placate the feminists, Ten Stupid Things Men Do at the other. She listens faithfully to Dr. Laura's daily denunciations of codependence, moral indecision, working mothers and drug-puffing teens. "Instant therapy," Gwen calls her show.
Of course, she never actually follows Dr. Laura's advice. That would be taking things to an extreme. No, Gwen is quite content remaining, like Ally, perpetually unhappy; she's addicted to The English Patient and to angst-ridden Ally McBeal list-servs. She complains of having lost her "passion" for religion. She repeats relational mistakes dozens of times, then spends Kleenex-filled evenings rereading old journals. How could she have time to pursue a healthy relationship, a spiritual life or meaningful work? Thousands of eighth grade diary pages are still waiting to be deconstructed!
Every so often I experience doubts about the implications of postmodern navel gazing. Periodically I wake to looming questions I would rather let Scully--or maybe a licensed ethicist--address. What are the moral, social and political consequences of an over-examined life? Can a person care responsibly for his or her "self" without becoming solipsistic and without abandoning responsibility to the common or public good? Does Generation X need an ethic of self-reflection?
One could argue, after all, that Dr. Laura's tactics for self-reflection are highly unethical. She interrupts working mothers with unjustified accusations of greed and selfishness. She applies careless verbal Band-Aids to gaping psychological and social wounds. She doesn't listen; she simply swoops in for the therapeutic kill.
One could also argue that Dr. Laura is as myopic as her waifish opposite. Ally's television show is a celebration of destructive behavior; Dr. Laura's radio show is a two-second "solution" to lifelong patterns of destructive behavior. Neither program bothers to distinguish between damaging versus constructive forms of self-examination. Neither pauses to consider the consequences of representing ethical and psychological lethargy as entertainment.
Thankfully, however, I have discovered that coffee and cigarettes provide an excellent antidote to such bothersome late-night misgivings. Armed with mug and cancer stick, I can proclaim to my rapt stuffed animals that while solipsism, social responsibility and myopic stagnation might be worth addressing, I personally do not have the time or luxury to look into any of them.
This is because I am busy perfecting my use of the navel gazer's favorite academic term: "paradox." Paradox is a good word because it allows us to be as contradictory as possible without having to explain our actions or justify a lack of personal ethics. For example:
Question: "But Stacia, how can you applaud Dr. Laura's absolutist moralistic tone while at the same time wallowing in Ally's haze of moral and personal ambiguity?" Answer: "It's a paradox!"
See how handy the word is? Thank God for higher education.
Navel gazers, take heart: Contrary to what that awful therapist once told you, being pathologically unhappy or perpetually self-destructive is not a bad thing. Our supposed flaws are actually rare gifts: They not only provide poetic distraction from healthy relationships and from life's deepest questions, they also make terrific diet aids. Just look at Ally: She's so busy chewing on her own personal drama, her body has disappeared almost entirely.
Now that's what I call self-absorption.
Stacia Brown is program associate at the Center for Ethics.
Return to April 5, 1999, contents page
```
.
TRC ASTS Abstracts
12/19/22
1. Too much text
2. Excel figures
3. Very poor quality
4. Table 1 + follow-up visits!
5. Incroyable!
.
Chappell says [this](https://en.wikipedia.org/wiki/Iceberg_Slim) is a metaphor of what he went through before surprise retirement
.
Swim
12/20/22
4x100m
3:03:03 (WR) = 183s
.
46/100m
23/50m
12.4/25m
.
```
Para todo mal,
mezcal, y
para todo bien,
también; y
si no
hay remedio
litro
y medio"
Suffers in translation:
For all bad,
mezcal, and
for all good,
as well; and
if there is
no remedy,
liter and
a half
```
.
autobiography
12/22/22
inspired by alfonso cuarón’s roma:
0. incestuous potential: family, neighbors
1. designing wire cars & fùtbols
2. morris cerullo & festo kivengere
3. heaven bound & breath again
4. s3 australia house party & s4 social
5. a-level biology with mubiro & music with katuramu
6. rugby nation-wide champions, mvp
7. homoerotic potential: effeminate subordinate, was intro to sheba
8. college of medicine &, finally, chaos in my life!
9. impis & a return to some order
10. onset of 20-years of décadence
11. enter hamlet, good by achilles
12. graduate medical school, ihk
13. should have flunked microbiology & medicine
14. public health as prudence: proverbs 27:12
15. numbers, statistics, bayes, philosophy, medicine in nutshell
16. it’s been quite an ache… 2004-2022
17. back-at-one (pathos, logos, ethos or nutshell)
.
```
Viagra (Soldenafil): Case study in serendipity and blunders
12/21/22
Viagra was developed and studied in phase I clinical trials for hypertension and angina pectoris (1). It’s therapeutic effects were not substantive and did not reach phase II or III trials. But a commonly reported side effect in the phase I trial was the induction of marked penile erections (2).
Pfizer immediately recognized its commercial potential and patented it in 1996. It was then repositioned for erectile dysfunction and was approved for erectile dysfunction by FDA within a mere two years(3).
Within 3 months of FDA licensing in 1998 (4), Pfizer had earned $400m from viagra and over the next 20 years leading to expiry of its patent in 2020 the annual earnings from the sale of viagra were $1.2 - $2.0b, with a cumulative total of $27.6b as of 2020 (5).
The market for viagra and its competitors is expected to continue growing according to the WHO. Increased burden of lifestyle disease and sedentary lifestyle interact to cause erectile dysfunction. At a prevalence of 15% of the male population, it is projected by WHO to reach 320m persons by 2025 (6). Such is the story of an entirely serendipitous discovery by otherwise astute scientists and clinicians that were meticulous enough to profit from unintended consequences.
References
1.History of sildenafil. Wikipedia. Accessed December 22, 2022. https://en.wikipedia.org/wiki/Sildenafil#History
2.Goldstein I, et al. The Serendipitous story of sildenafil: an unexpected oral therapy for erectile dysfunction. ScienceDirect. Volume 7, Issue 1, January 2019, Pages 115-128
3.Ellis P, et al. Pyrazolopyrimidinones for the treatment of impotence. Google Patents. Accessed December 22, 2022. https://patents.google.com/patent/US6469012B1/en
4. Cox, D. The race to replace viagra. The Guardian. Accessed https://www.theguardian.com/science/2019/jun/09/race-to-replace-viagra-patents-erectile-dysfunction-drug-medical-research-cialis-eroxon
5.Worldwide revenue of Pfizer’s viagra from 2003 to 2019. Statista. Accessed December 22, 2022. https://www.statista.com/statistics/264827/pfizers-worldwide-viagra-revenue-since-2003/
6..Erectile dysfunction drugs market size, share & trends analysis report by product (viagra, Cialis, zydeco, levity, stendra), by region (North America, Europe, APAC, Latin America, MEA), and segment forecasts, 2022-2030. Grand View Research. Accessed December 22, 2022. https://www.grandviewresearch.com/industry-analysis/erectile-dysfunction-drugs-market
```
.
Phrase of the year
12/25/22
He thinks in terms of profit & loss not virtue & vice, which is just as well given that he wears flip flops and has a solar panel attached to his thatched roof, breaking the PROHIBITIONS against shoes & and electricity
.
Kardamon & Kofee
Otters bar
Ruhanga Tayebwa (God didn’t forget)
.
Okusumagira — Esau
Instead of condemning the blinker, praise the fraud by
.
Yanungamya
|
{
"filename": "bdn2022.ipynb",
"repository": "muzaale/denotas",
"query": "transformed_from_existing",
"size": 300281,
"sha": ""
}
|
# process_gre_output_1.ipynb
Repository: pat-jj/GenRES
<code>
import json
filepath_gpt4_turbo = 'results/wiki20m_rand_100_gpt-4-1106-preview_detailed.json'
filepath_gt = 'results/wiki20m_rand_100_groundtruth_detailed.json'
filepath_llama2 = 'results/wiki20m_rand_100_llama-2-70b_detailed.json'
filepath_openchat = 'results/wiki20m_rand_100_openchat_detailed.json'
with open(filepath_gpt4_turbo) as f:
gpt4_turbo = json.load(f)
with open(filepath_gt) as f:
gt = json.load(f)
with open(filepath_llama2) as f:
llama2 = json.load(f)
with open(filepath_openchat) as f:
openchat = json.load(f)
</code>
<code>
sample_to_id = {}
for idx, key in enumerate(gpt4_turbo.keys()):
sample_to_id[key] = idx
sample_to_id
</code>
<code>
import pandas as pd
metrics_names = ['Completeness', 'Factualness', 'Granularity', 'Topical', 'Uniqueness']
metrics_names_to_abbr = {
'Completeness': 'CS',
'Factualness': 'FS',
'Granularity': 'GS',
'Topical': 'TS',
'Uniqueness': 'US'
}
gre_results_dict = {}
for metric_name in metrics_names:
df = pd.DataFrame(columns=["sample_id", "Groundtruth", "LLaMA2-70b", "OpenChat", "GPT-4-Turbo"])
metric = metrics_names_to_abbr[metric_name]
for key in sample_to_id.keys():
sample_id = sample_to_id[key]
gt_score = gt[key][metric]
llama2_score = llama2[key][metric]
openchat_score = openchat[key][metric]
gpt4_turbo_score = gpt4_turbo[key][metric]
comparison = {
"sample_id": sample_id,
"Groundtruth": gt_score,
"LLaMA2-70b": llama2_score,
"OpenChat": openchat_score,
"GPT-4-Turbo": gpt4_turbo_score
}
df = pd.concat([df, pd.DataFrame([comparison])], ignore_index=True)
gre_results_dict[metric_name] = df
</code>
<code>
gre_results_dict['Completeness']
</code>
<code>
import os
# Function to create pairwise comparisons between models
def create_pairwise_comparisons(df):
# List of models for comparison
model_columns = df.columns[1:] # Excluding the sample ID column
# Create an empty DataFrame for the pairwise comparisons
comparisons_df = pd.DataFrame(columns=["sample_id", "model_A_name", "model_B_name", "win"])
threshold = 0.2
# Iterate through each row in the DataFrame
for _, row in df.iterrows():
# Get the sample ID
sample_id = row.iloc[0]
# Compare each model with every other model
for i in range(len(model_columns)):
for j in range(i+1, len(model_columns)):
model_a = model_columns[i]
model_b = model_columns[j]
scores_a = row[model_a]
scores_b = row[model_b]
# Determine the winner
if scores_a - scores_b >= threshold:
win = 'model_A_win'
elif scores_b - scores_a >= threshold :
win = 'model_B_win'
else:
win = 'tie'
# Add the comparison to the DataFrame
comparison = {"sample_id": sample_id, "model_A_name": model_a, "model_B_name": model_b, "win": win}
comparisons_df = pd.concat([comparisons_df, pd.DataFrame([comparison])], ignore_index=True)
return comparisons_df
</code>
<code>
for metric_name in metrics_names:
df = gre_results_dict[metric_name]
pairwise_comparisons_df = create_pairwise_comparisons(df)
# obtain the filename
csv_filename = f'{metric_name}_GREScores.csv'
pairwise_comparisons_df.to_csv(os.path.join('results/gre', csv_filename), index=False)
</code>
|
{
"filename": "process_gre_output_1.ipynb",
"repository": "pat-jj/GenRES",
"query": "transformed_from_existing",
"size": 28526,
"sha": ""
}
|
# ChIP_TIP.ipynb
Repository: gersteinlab/LatentDAG
<code>
import os
import pandas as pd
import scanpy as sc
</code>
<code>
genes = pd.read_csv("../../result/network_perturb_go/valid_genes", sep="\t")
id2genes = genes.set_index("ID")["genes"].to_dict()
genes2id = genes.set_index("genes")["ID"].to_dict()
genes = genes['genes'].values
</code>
### preprocess
<code>
# # filter valid experiments from ENCODE-download metadata
# meta = pd.read_csv("../../data/TIP/metadata.tsv", sep="\t")
# meta['TF'] = meta['Experiment target'].str.split("-human", expand=True)[0]
# meta = meta[meta['TF'].isin(genes)] # keep TF in genes only
# meta = meta[meta['Output type'] == "IDR thresholded peaks"]
# meta.iloc[:, [0, -1]].to_csv("../../data/TIP/meta_perturb.tsv", sep="\t", index=False)
# # download ChIP-seq bed files
# with open("joblist", "w") as ff:
# for fid in meta['File accession'].values:
# if not os.path.exists("../../data/TIP/ENCODE/%s.bed.gz" % fid):
# ff.write("wget https://www.encodeproject.org/files/%s/@@download/%s.bed.gz\n" % (fid, fid))
# # move downloaded files to ../../data/TIP/ENCODE/
# # run TIP
# meta = pd.read_csv("../../data/TIP/meta_perturb.tsv", sep="\t")
# with open("joblist", "w") as f:
# for fid in meta["File accession"]:
# f.write("module load R; Rscript TIP.R ../../data/TIP/gene_annotation.txt ../../data/TIP/ENCODE/%s.bed.gz ../../data/TIP/weight/%s.txt ../../data/TIP/score/%s.txt\n" % (fid, fid, fid))
# # calculate q-value
# meta = pd.read_csv("../../data/TIP/meta_perturb.tsv", sep="\t")
# with open("joblist", "w") as f:
# for fid in meta["File accession"]:
# f.write("module load R; Rscript TIP_qval.R ../../data/TIP/score/%s.txt ../../data/TIP/qval/%s.txt\n" % (fid, fid))
</code>
### parse result to network
<code>
meta = pd.read_csv("../../data/TIP/meta_perturb.tsv", sep="\t")
</code>
<code>
data = sc.read_h5ad("../../data/perturb/K562_gwps_normalized_bulk_01.h5ad")
eid2name = data.var[data.var['gene_name'].isin(genes)]['gene_name'].to_dict()
result = {}
for fid, TF in meta.values:
df = pd.read_csv("../../data/TIP/qval/%s.txt" % fid, sep="\t")
df["name"] = df["name"].map(eid2name) # keep target in genes only and convert to gene name
df = df.dropna()
targets = set(df['name'].unique())
if TF in result.keys():
result[TF] = set.union(result[TF], targets)
else:
result[TF] = targets
</code>
<code>
source = []
target = []
for ii in result.keys():
source += [ii] * len(result[ii])
target += list(result[ii])
</code>
<code>
net = pd.DataFrame([source, target]).T
net[0] = net[0].map(genes2id)
net[1] = net[1].map(genes2id)
net = net.dropna().copy() # remove any NA
net[0] = net[0].astype(int)
net[1] = net[1].astype(int)
</code>
<code>
net = net.drop_duplicates().sort_values([0, 1])
net.to_csv("../../result/network_perturb_go/ChIP_TIP.tsv", sep="\t", header=False, index=False)
</code>
|
{
"filename": "ChIP_TIP.ipynb",
"repository": "gersteinlab/LatentDAG",
"query": "transformed_from_existing",
"size": 5707,
"sha": ""
}
|
# to_Python_Deitel_01_13.ipynb
Repository: weigeng-valpo/Intro
# 1.13 How Big Is Big Data?
For computer scientists and data scientists, data is now as important as writing programs
* According to IBM, approximately 2.5 quintillion bytes (2.5 _exabytes_) of data are created daily, and 90% of the world’s data was created in the last two years
* According to IDC, the global data supply will reach 175 _zettabytes_ (equal to 175 trillion gigabytes or 175 billion terabytes) annually by 2025
### Megabytes (MB)
* One megabyte is about one million (actually 2<sup>20</sup>) bytes
* Many of the files we use on a daily basis require one or more MBs of storage
* MP3 audio files—High-quality MP3s range from 1 to 2.4 MB per minute
* Photos—JPEG format photos taken on a digital camera can require about 8 to 10 MB per photo
* Video—Smartphone cameras can record video at various resolutions
* Each minute of video can require many megabytes of storage
* On one of our iPhones, the **Camera** settings app reports that 1080p video at 30 frames-per-second (FPS) requires 130 MB/minute and 4K video at 30 FPS requires 350 MB/minute
### Gigabytes (GB)
* One gigabyte is about 1000 megabytes (actually 2<sup>30</sup> bytes
* A dual-layer DVD can store up to 8.5 GB, which translates to:
* as much as 141 hours of MP3 audio
* approximately 1000 photos from a 16-megapixel camera
* approximately 7.7 minutes of 1080p video at 30 FPS
* approximately 2.85 minutes of 4K video at 30 FPS
* Highest-capacity Ultra HD Blu-ray discs can store up to 100 GB of video
* Streaming a 4K movie can use between 7 and 10 GB per hour (highly compressed)
### Terabytes (TB)
* One terabyte is about 1000 gigabytes (actually 2<sup>40</sup> bytes)
* Recent disk drives for desktop computers come in sizes up to 15 TB, which is equivalent to
* approximately 28 years of MP3 audio
* approximately 1.68 million photos from a 16-megapixel camera
* approximately 226 hours of 1080p video at 30 FPS
* approximately 84 hours of 4K video at 30 FPS
* Nimbus Data now has the largest solid-state drive (SSD) at 100 TB, which can store 6.67 times the 15-TB examples of audio, photos and video listed above
### Petabytes, Exabytes and Zettabytes
* There are nearly four billion people online creating about 2.5 quintillion bytes of data each day
* 2500 petabytes (each petabyte is about 1000 terabytes) or 2.5 exabytes (each exabyte is about 1000 petabytes)
* According to a March 2016 _AnalyticsWeek_ article, within five years there will be over 50 billion devices connected to the Internet and by 2020 we’ll be producing 1.7 megabytes of new data every second _for every person on the planet_
* At today’s numbers (approximately 7.7 billion people), that’s about
* 13 petabytes of new data per second
* 780 petabytes per minute
* 46,800 petabytes (46.8 exabytes) per hour
* 1,123 exabytes per day—that’s 1.123 zettabytes (ZB) per day (each zettabyte is about 1000 exabytes)
* That’s the equivalent of over 5.5 million hours (over 600 years) of 4K video every day or approximately 116 billion photos every day!
### Additional Big-Data Stats
* For an entertaining real-time sense of big data, check out https://www.internetlivestats.com, with various statistics, including the numbers so far today of
* Google searches
* Tweets
* Videos viewed on YouTube
* Photos uploaded on Instagram
### Additional Big-Data Stats (cont.)
* Every hour, YouTube users upload 24,000 hours of video, and almost 1 billion hours of video are watched on YouTube every day
* Every second, there are 51,773 GBs (or 51.773 TBs) of Internet traffic, 7894 tweets sent, 64,332 Google searches and 72,029 YouTube videos viewed
* On Facebook each day there are 800 million “**likes**,” 60 million emojis are sent, and there are over two billion searches of the more than 2.5 trillion Facebook posts since the site’s inception
### Additional Big-Data Stats (cont.)
* In June 2017, Will Marshall, CEO of Planet, said the company has 142 satellites that image the whole planet’s land mass once per day
* They add one million images and seven TBs of new data each day
* They’re using machine learning on that data to improve crop yields, see how many ships are in a given port and track deforestation
* With respect to Amazon deforestation, he said: “Used to be we’d wake up after a few years and there’s a big hole in the Amazon. Now we can literally count every tree on the planet every day.”
### Additional Big-Data Stats (cont.)
Domo, Inc. has a nice infographic called “Data Never Sleeps 6.0” showing how much data is generated _every minute_, including:
* 473,400 tweets sent.
* 2,083,333 Snapchat photos shared.
* 97,222 hours of Netflix video viewed.
* 12,986,111 million text messages sent.
* 49,380 Instagram posts.
* 176,220 Skype calls.
* 750,000 Spotify songs streamed.
* 3,877,140 Google searches.
* 4,333,560 YouTube videos watched.
### Computing Power Over the Years
* Data is getting more massive and so is the computing power for processing it
* Performance of today’s processors is measured in terms of **FLOPS (floating-point operations per second)**
* In the early to mid-1990s, the fastest supercomputer speeds were measured in gigaflops (109 FLOPS)
* Late 1990s: Intel produced the first teraflop (10<sup>12</sup> FLOPS) supercomputers
* Early-to-mid 2000s: Speeds reached hundreds of teraflops
* 2008: IBM released the first petaflop (10<sup>15</sup> FLOPS) supercomputer
* Currently, the fastest supercomputer—the IBM Summit, located at the Department of Energy’s (DOE) Oak Ridge National Laboratory (ORNL)—is capable of 122.3 petaflops
### Computing Power Over the Years (cont.)
* Distributed computing can link thousands of personal computers via the Internet to produce even more FLOPS
* 2016: The Folding@home network—a distributed network in which people volunteer their personal computers’ resources for use in disease research and drug design—was capable of over 100 petaflops
* Companies like IBM are now working toward supercomputers capable of exaflops (10<sup>18</sup> FLOPS)
### Computing Power Over the Years (cont.)
* **Quantum computers** now under development theoretically could operate at 18,000,000,000,000,000,000 times the speed of today’s “conventional computers”!
* In one second, a quantum computer theoretically could do staggeringly more calculations than the total that have been done by all computers since the world’s first computer appeared.
* Could wreak havoc with blockchain-based cryptocurrencies like Bitcoin
* Engineers are already rethinking blockchain to prepare for such massive increases in computing power
### Computing Power Over the Years (cont.)
* Computing power’s cost continues to decline, especially with cloud computing
* People used to ask the question, “How much computing power do I need on my system to deal with my _peak_ processing needs?”
* That thinking has shifted to “Can I quickly carve out on the cloud what I need _temporarily_ for my most demanding computing chores?”
* Pay for only what you use to accomplish a given task
### Processing the World’s Data Requires Lots of Electricity
* Data from the world’s Internet-connected devices is exploding, and processing that data requires tremendous amounts of energy.
* According to a recent article, energy use for processing data in 2015 was growing at 20% per year and consuming approximately three to five percent of the world’s power
* That total data-processing power consumption could reach 20% by 2025
### Processing the World’s Data Requires Lots of Electricity (cont.)
* Another enormous electricity consumer is the blockchain-based cryptocurrency Bitcoin
* Processing just one Bitcoin transaction uses approximately the same amount of energy as powering the average American home for a week!
* The energy use comes from the process Bitcoin “miners” use to prove that transaction data is valid
### Big-Data Opportunities
* Big data’s appeal to big business is undeniable given the rapidly accelerating accomplishments
* Many companies are making significant investments and getting valuable results through technologies in this book, such as big data, machine learning, deep learning and natural-language processing
* Forcing competitors to invest as well, rapidly increasing the need for computing professionals with data-science and computer science experience
## 1.13.1 Big Data Analytics
* The term “data analysis” was coined in 1962, though people have been analyzing data using statistics for thousands of years going back to the ancient Egyptians
* Big data analytics is a more recent phenomenon—the term “big data” was coined around 2000
* Four of the V’s of big data:
1. Volume—the amount of data the world is producing is growing exponentially.
2. Velocity—the speed at which that data is being produced, the speed at which it moves through organizations and the speed at which data changes are growing quickly.
3. Variety—data used to be alphanumeric (that is, consisting of alphabetic characters, digits, punctuation and some special characters)—today it also includes images, audios, videos and data from an exploding number of Internet of Things sensors in our homes, businesses, vehicles, cities and more.
4. Veracity—the validity of the data—is it complete and accurate? Can we trust that data when making crucial decisions? Is it real?
## 1.13.1 Big Data Analytics (cont.)
* Most data is now being created digitally in a _variety_ of types, in extraordinary _volumes_ and moving at astonishing _velocities_
* Digital data storage has become so vast in capacity, cheap and small that we can now conveniently and economically retain _all_ the digital data we’re creating
## 1.13.1 Big Data Analytics (cont.)
To get a sense of big data’s scope in industry, government and academia, check out the high-resolution graphic
> http://mattturck.com/wp-content/uploads/2018/07/Matt_Turck_FirstMark_Big_Data_Landscape_2018_Final.png
## 1.13.2 Data Science and Big Data Are Making a Difference: Use Cases
* The data-science field is growing rapidly because it’s producing significant results that are making a difference
* Some data-science and big data use cases in the following table
| Data-science use cases
| ------------
| anomaly detection
| assisting people with disabilities
| auto-insurance risk prediction
| automated closed captioning
| automated image captions
| automated investing
| autonomous ships
| brain mapping
| caller identification
| cancer diagnosis/treatment
| carbon emissions reduction
| classifying handwriting
| computer vision
| credit scoring
| crime: predicting locations
| crime: predicting recidivism
| crime: predictive policing
| crime: prevention
| CRISPR gene editing
| crop-yield improvement
| customer churn
| customer experience
| customer retention
| customer satisfaction
| customer service
| customer service agents
| customized diets
| cybersecurity
| data mining
| data visualization
| detecting new viruses
| diagnosing breast cancer
| diagnosing heart disease
| diagnostic medicine
| disaster-victim identification
| drones
| dynamic driving routes
| dynamic pricing
| electronic health records
| emotion detection
| energy-consumption reduction
| facial recognition
| fitness tracking
| fraud detection
| game playing
| genomics and healthcare
| Geographic Information Systems (GIS)
| GPS Systems
| health outcome improvement
| hospital readmission reduction
| human genome sequencing
| identity-theft prevention
| immunotherapy
| insurance pricing
| intelligent assistants
| Internet of Things (IoT) and medical device monitoring
| Internet of Things and weather forecasting
| inventory control
| language translation
| location-based services
| loyalty programs
| malware detection
| mapping
| marketing
| marketing analytics
| music generation
| natural-language translation
| new pharmaceuticals
| opioid abuse prevention
| personal assistants
| personalized medicine
| personalized shopping
| phishing elimination
| pollution reduction
| precision medicine
| predicting cancer survival
| predicting disease outbreaks
| predicting health outcomes
| predicting student enrollments
| predicting weather-sensitive product sales
| predictive analytics
| preventative medicine
| preventing disease outbreaks
| reading sign language
| real-estate valuation
| recommendation systems
| reducing overbooking
| ride sharing
| risk minimization
| robo financial advisors
| security enhancements
| self-driving cars
| sentiment analysis
| sharing economy
| similarity detection
| smart cities
| smart homes
| smart meters
| smart thermostats
| smart traffic control
| social analytics
| social graph analysis
| spam detection
| spatial data analysis
| sports recruiting and coaching
| stock market forecasting
| student performance assessment
| summarizing text
| telemedicine
| terrorist attack prevention
| theft prevention
| travel recommendations
| trend spotting
| visual product search
| voice recognition
| voice search
| weather forecasting
------
©1992–2020 by Pearson Education, Inc. All Rights Reserved. This content is based on Chapter 1 of the book [**Intro to Python for Computer Science and Data Science: Learning to Program with AI, Big Data and the Cloud**](https://amzn.to/2VvdnxE).
DISCLAIMER: The authors and publisher of this book have used their
best efforts in preparing the book. These efforts include the
development, research, and testing of the theories and programs
to determine their effectiveness. The authors and publisher make
no warranty of any kind, expressed or implied, with regard to these
programs or to the documentation contained in these books. The authors
and publisher shall not be liable in any event for incidental or
consequential damages in connection with, or arising out of, the
furnishing, performance, or use of these programs.
|
{
"filename": "to_Python_Deitel_01_13.ipynb",
"repository": "weigeng-valpo/Intro",
"query": "transformed_from_existing",
"size": 19770,
"sha": ""
}
|
# bioimage_analysis_course_2025_day1_1_instant_gratification_1.ipynb
Repository: brunicardoso/python
# **Instant Gratification**
**Install required packages and specific modules from packages**
<code>
from skimage.io import imread, imsave
import numpy as np
import matplotlib.pyplot as plt
from matplotlib.patches import Rectangle
</code>
**Download image**
image source https://imagej.net/ij/images/ . This is an example dataset from ImageJ
<code>
!wget https://imagej.net/ij/images/3_channel_inverted_luts.tif
</code>
**Visualize image**
<code>
img= imread('3_channel_inverted_luts.tif')
plt.imshow(img[0:708,0:708:,0], cmap='grey')
plt.axis("off")
</code>
# **It is your turn to try out different things**
1. Inspect the image by running the code img.shape. The first two values are y and x pixel dimensions and the third value is the number of channels.
This order might be different for different images. Sometimes the 'channel' dimension comes first.
2. This image has 4 channels. Modify the code plt.imshow(img[:,:,0], cmap='grey') to visualize the other channels.
3. Play around with the color map (cmap). Check out other cmap options here https://matplotlib.org/stable/users/explain/colors/colormaps.html
4. Crop the image by limiting the pixel range being displayed. For example plt.imshow(img[200:400,200:400:,0], cmap='grey')
5. Test plt.imshow(img[:,:,:,], cmap='grey') and plt.imshow(img[:,:,0], cmap='grey')
6. Add the hashtag # symbol before the code and try to run it. What happens?
6. Download another image from the internet and play with it.
# **Images are numbers**
Images are composed of arrays of numbers (tables). Because the image we are working on is quite large (708 by 708 pixels), the full table of numbers can not be displayed
<code>
print(img)
</code>
Printing fewer pixels give us a better idea of the images are numbers concept
<code>
print(img[340:350, 340:350,0])
</code>
<code>
plt.imshow(img[340:350, 340:350,0], cmap='grey')
plt.axis("off")
</code>
**Our image is an 16 bit image, thus each pixel can have a value up to 65536 (2^16). A pixel value of 0 (the least intense) is black whereas a pixel value of 65535 (the most intense) is white** .
If you want to learn more abot bit depth, here a good start. https://bioimagebook.github.io/chapters/1-concepts/3-bit_depths/bit_depths.html
<code>
# @title
import numpy as np
import matplotlib.pyplot as plt
from matplotlib.patches import Rectangle
original_image = img[:, :, 0]
image = img[:, :, 0]
# Define rectangle coordinates
start_row, end_row = 340, 350
start_col, end_col = 340, 350
width, height = end_col - start_col, end_row - start_row
# Create subplots
fig, axs = plt.subplots(1, 3, figsize=(15, 15))
# First subplot: original image with rectangle
axs[0].imshow(original_image, cmap='gray', interpolation='nearest')
rect = Rectangle(
(start_col, start_row), width, height,
edgecolor='blue', facecolor='none', linewidth=1.5
)
axs[0].add_patch(rect)
axs[0].set_title("Original Image with Rectangle")
# Second subplot: zoomed-in region without text
axs[1].imshow(img[start_row:end_row, start_col:end_col, 0], cmap='gray', interpolation='nearest')
axs[1].set_title("Zoomed-In Region")
# Third subplot: zoomed-in region with pixel values
zoomed_image = img[start_row:end_row, start_col:end_col, 0]
axs[2].imshow(zoomed_image, cmap='gray', interpolation='nearest')
# Overlay pixel values on the zoomed-in region
rows, cols = zoomed_image.shape
for i in range(rows):
for j in range(cols):
axs[2].text(j, i, f"{int(zoomed_image[i, j])}", color='white', ha='center', va='center', fontsize=8)
axs[2].set_title("Zoomed-In Region with Pixel Values")
# Remove axis ticks
for ax in axs:
ax.axis("off")
plt.tight_layout()
plt.show()
</code>
The image below is an 8 bit image, thus each pixel can have a value up to 256 (2^8). A pixel value of 0 (the least intense) is black whereas a pixel value of 255 (the most intense) is white
# **Its your turn**
1. Display array as an image
2. Change values in the array and visualize the changes
<code>
array = np.array(
[[255, 20, 0, 255, 20, 2], [255, 110, 0, 255, 20, 2], [255, 0, 0, 255, 20, 2],
[255, 20, 0, 255, 20, 2], [255, 67, 0, 255, 20, 2],[255, 0, 0, 255, 20, 2]]
)
print(array)
</code>
<code>
img=1
</code>
|
{
"filename": "bioimage_analysis_course_2025_day1_1_instant_gratification_1.ipynb",
"repository": "brunicardoso/python",
"query": "transformed_from_existing",
"size": 9181,
"sha": ""
}
|
# trials_matching.ipynb
Repository: nigat12/ai-treatment-connect
<code>
%pip install openpyxl
</code>
<code>
# --- Setup and Imports ---
import pandas as pd
import numpy as np
from sentence_transformers import SentenceTransformer
from sklearn.metrics.pairwise import cosine_similarity
import re # For cleaning text
import math # For isnan
</code>
<code>
# --- Configuration ---
XLSX_FILENAME = 'trials.xlsx' # Make sure your file is named this or change here
# Columns containing text descriptions relevant for semantic matching
# Conditions is the primary target as requested
TEXT_COLUMNS_FOR_EMBEDDING = ['Conditions'] # Adding Brief Summary can provide context
# You could potentially add 'Interventions' or 'Study Title' too depending on desired specificity
# Columns and criteria for structured filtering
FILTER_PRIMARY_OUTCOME_COLUMN = 'Primary Outcome Measures'
FILTER_PRIMARY_OUTCOME_TERM = 'Overall Survival'
FILTER_PHASES_COLUMN = 'Phases'
# Note: Splitting Phase combinations for checking is safer.
# Example: 'PHASE2|PHASE3' should match if user wants PHASE2 or PHASE3
ACCEPTABLE_PHASES = ['PHASE1|PHASE2', 'PHASE2', 'PHASE2|PHASE3', 'PHASE3', 'PHASE4']
ACCEPTABLE_INDIVIDUAL_PHASES = ACCEPTABLE_PHASES
# Let's create a set of individual phases for flexible checking
# ACCEPTABLE_INDIVIDUAL_PHASES = set()
# for phase_combo in ACCEPTABLE_PHASES:
# for phase in phase_combo.split('|'):
# ACCEPTABLE_INDIVIDUAL_PHASES.add(phase.strip())
FILTER_STUDY_TYPE_COLUMN = 'Study Type'
FILTER_STUDY_TYPE_VALUE = 'INTERVENTIONAL'
# Relevance score threshold (semantic similarity).
# Only trials with a semantic similarity score AT or ABOVE this threshold will be considered relevant.
# Needs tuning. Medical text similarity can be lower than general text for related concepts.
# Start with a value, evaluate results, and adjust.
RELEVANCE_SCORE_THRESHOLD = 0.5 # Slightly lower threshold might be needed for trial text variability
</code>
<code>
# --- Helper Functions ---
def clean_text(text):
"""Basic text cleaning: lowercase, remove special characters except hyphen, handle spaces."""
if isinstance(text, str):
text = text.lower()
# Keep letters, numbers, spaces, hyphens, and maybe some punctuation like comma/slash useful in medical text?
text = re.sub(r'[^a-z0-9\s-/]', '', text) # Added / for common phase notations like I/II
text = re.sub(r'\s+', ' ', text).strip() # Remove extra spaces
return text
return ''
# Helper for sorting with None/NaN values (None/NaN goes to the end)
def sort_key_with_none(value, reverse=True):
if value is None or (isinstance(value, float) and math.isnan(value)):
return float('-inf') if reverse else float('inf')
return value
# Function to check if a trial's phases match the acceptable list
def check_phases(trial_phases_raw):
if not isinstance(trial_phases_raw, str):
return False
trial_phases_cleaned = trial_phases_raw
# Split the trial's phase string by | or space and check if any individual phase is in our acceptable set
trial_individual_phases = re.split(r'[|\s]+', trial_phases_cleaned)
# Check if *any* phase mentioned in the trial is in our list of acceptable *individual* phases
# This makes the filter more flexible (e.g., 'PHASE2|PHASE3' passes if we accept PHASE2 or PHASE3)
# If you require the *exact* combination to be in ACCEPTABLE_PHASES list, simplify this logic.
# Assuming flexibility is better for matching.
for phase in trial_individual_phases:
if phase.upper() in [p.replace('|', '/') for p in ACCEPTABLE_PHASES] or phase.upper().replace('/', '|') in ACCEPTABLE_PHASES:
# Handle cases like 'PHASE III' vs 'PHASE3' - clean_text helps, but maybe add a map?
# Let's rely on clean_text and upper() for now.
# Direct check against the original ACCEPTABLE_PHASES list:
if trial_phases_raw.upper() in ACCEPTABLE_PHASES:
return True
# Check individual components against ACCEPTABLE_INDIVIDUAL_PHASES
if phase.upper() in ACCEPTABLE_INDIVIDUAL_PHASES:
return True
return False
</code>
<code>
# --- Data Loading and Preprocessing ---
print(f"Loading data from {XLSX_FILENAME}...")
try:
# Use pd.read_excel for .xlsx files
df = pd.read_excel(XLSX_FILENAME)
print("Data loaded successfully.")
print(f"Initial data shape: {df.shape}")
except FileNotFoundError:
print(f"Error: {XLSX_FILENAME} not found. Please make sure the XLSX file is in the same directory.")
exit()
# Preprocess text columns for embedding
# Combine relevant text columns into a single column
# Ensure columns exist before accessing
text_cols_present = [col for col in TEXT_COLUMNS_FOR_EMBEDDING if col in df.columns]
if not text_cols_present:
print(f"Error: None of the specified text columns for embedding ({TEXT_COLUMNS_FOR_EMBEDDING}) found in the file.")
exit()
# print(df[text_cols_present].head())
df['combined_text_for_embedding'] = df[text_cols_present[0]].fillna('').astype(str)
# df['combined_text_for_embedding'] = df[text_cols_present].fillna('').agg(' '.join, axis=1)
df['combined_text_cleaned_for_embedding'] = df['combined_text_for_embedding']
</code>
<code>
# Load Medical Domain Sentence Transformer model
print("\nLoading Medical Domain Sentence Transformer model...")
try:
# Using PubMedBERT finetuned for sentence similarity
model = SentenceTransformer('all-MiniLM-L6-v2')
print("Model loaded successfully.")
except Exception as e:
print(f"Error loading Sentence Transformer model: {e}")
print("Please ensure you have internet access or the model files are cached, or try a different model name.")
exit()
# Generate embeddings for the combined text (done once for the whole dataset)
print("\nGenerating text embeddings for trial data...")
# Only generate embeddings for rows where the text column is not empty after cleaning
# This saves computation and avoids issues with empty strings in some models
non_empty_indices = df.index[df['combined_text_cleaned_for_embedding'].str.strip() != ''].tolist()
non_empty_texts = df.loc[non_empty_indices, 'combined_text_cleaned_for_embedding'].tolist()
if not non_empty_texts:
print("Warning: No non-empty text found in the specified columns for embedding. Cannot generate embeddings.")
drug_embeddings = np.array([]) # Create empty array to avoid errors
else:
drug_embeddings = model.encode(non_empty_texts, show_progress_bar=True, convert_to_numpy=True)
print(f"Embeddings generated for {len(non_empty_indices)} trials.")
# Create a mapping from original DataFrame index to embedding index
index_to_embedding_index = {original_idx: emb_idx for emb_idx, original_idx in enumerate(non_empty_indices)}
</code>
<code>
# --- Search Function ---
def find_relevant_trials(df: pd.DataFrame, drug_embeddings: np.ndarray, index_to_embedding_index: dict,
model: SentenceTransformer,
user_cancer_type_raw: str, user_stage_raw: str, user_biomarkers_raw: str,
relevance_threshold: float = RELEVANCE_SCORE_THRESHOLD):
"""
Finds and ranks relevant clinical trials based on user-provided cancer information,
structured filters, and semantic similarity.
Args:
df: The pre-processed DataFrame.
drug_embeddings: Pre-calculated embeddings for the trial data (non-empty texts).
index_to_embedding_index: Mapping from DataFrame index to embedding index.
model: The loaded Sentence Transformer model.
user_cancer_type_raw: The raw string input for cancer type.
user_stage_raw: The raw string input for stage.
user_biomarkers_raw: The raw string input for biomarkers (comma-separated).
relevance_threshold: The minimum semantic similarity score for a trial to be considered relevant.
Returns:
A list of dictionaries, each representing a relevant trial result with details and scores.
"""
# Clean user inputs
user_cancer_type_cleaned = user_cancer_type_raw
user_stage_cleaned = user_stage_raw
user_biomarkers_cleaned_list = [b.strip() for b in user_biomarkers_raw.split(',') if b.strip()]
# Create the full user query string for embedding
# Include all parts of the user's profile for semantic matching
user_query_text = f"{user_cancer_type_cleaned} {user_stage_cleaned} {' '.join(user_biomarkers_cleaned_list)}"
print(f"\n--- Searching for trials for profile: {user_query_text.strip()} ---")
if not user_query_text.strip():
print("Warning: User query is empty after cleaning. Cannot perform search.")
return []
# Generate embedding for the user query
try:
user_embedding = model.encode(user_query_text, convert_to_numpy=True)
except Exception as e:
print(f"Error generating user query embedding: {e}")
return []
potential_results = []
# Iterate through the pre-processed DataFrame
for index, row in df.iterrows():
# Skip rows that did not have embeddings generated
if index not in index_to_embedding_index:
continue
# --- Apply Structured Filters ---
# 1. Filter by Primary Outcome Measures
primary_outcome_text = str(row.get(FILTER_PRIMARY_OUTCOME_COLUMN, '')).lower()
if FILTER_PRIMARY_OUTCOME_TERM.lower() not in primary_outcome_text:
#print(f"Skipping trial {row.get('NCT Number', index)}: Did not match Primary Outcome filter.")
continue # Skip if Primary Outcome filter not met
# 2. Filter by Phases
trial_phases_raw = row.get(FILTER_PHASES_COLUMN, '')
if not check_phases(trial_phases_raw):
#print(f"Skipping trial {row.get('NCT Number', index)}: Did not match Phases filter (was '{trial_phases_raw}').")
continue # Skip if Phases filter not met
# 3. Filter by Study Type
trial_study_type = str(row.get(FILTER_STUDY_TYPE_COLUMN, '')).upper() # Assume uppercase for filter value
if trial_study_type != FILTER_STUDY_TYPE_VALUE.upper():
#print(f"Skipping trial {row.get('NCT Number', index)}: Did not match Study Type filter (was '{trial_study_type}').")
continue # Skip if Study Type filter not met
# --- Calculate Semantic Similarity (Only for trials passing filters) ---
embedding_index = index_to_embedding_index[index]
semantic_sim = cosine_similarity([user_embedding], [drug_embeddings[embedding_index]])[0][0]
# --- Filter by Relevance Threshold ---
if semantic_sim >= relevance_threshold:
# Store Result with Data and Scores if above threshold
potential_results.append({
'index': index,
'semantic_similarity': semantic_sim,
# Include relevant original data for display/explanation
'NCT Number': row.get('NCT Number', 'N/A'),
'Study Title': row.get('Study Title', 'N/A'),
'Study Status': row.get('Study Status', 'N/A'),
'Conditions': row.get('Conditions', 'N/A'),
'Interventions': row.get('Interventions', 'N/A'),
'Phases': row.get('Phases', 'N/A'),
'Brief Summary': row.get('Brief Summary', 'N/A'),
'Primary Outcome Measures': row.get('Primary Outcome Measures', 'N/A'),
})
# --- Ranking ---
# Sort results (which are already filtered):
# 1. Primarily by Semantic Similarity (descending)
# 2. Secondarily by Phase (later phases often more relevant clinical question)
# Let's assign a numerical value to phases for sorting: PHASE4 > PHASE3 > PHASE2|PHASE3 > PHASE2 > PHASE1|PHASE2
phase_order = {
'PHASE4': 5,
'PHASE3': 4,
'PHASE2|PHASE3': 3,
'PHASE2': 2,
'PHASE1|PHASE2': 1
}
def get_phase_sort_value(phases_raw):
if not isinstance(phases_raw, str): return 0 # Treat None/empty as lowest priority
phases_upper = phases_raw.upper()
return phase_order.get(phases_upper, 0) # Default to 0 if phase is not in our list
potential_results.sort(key=lambda x: (
x['semantic_similarity'], # Primary: Semantic Similarity (desc)
# get_phase_sort_value(x.get('Phases')), # Secondary: Phase (desc)
# x.get('Study Status', 'ZZZ') # Tertiary: Study Status (alphabetical, putting 'Recruiting' earlier)
), reverse=True) # Sort overall descending based on the tuple components
# --- Present Results ---
print(f"\nFound {len(potential_results)} relevant trials (Semantic Similarity >= {relevance_threshold:.2f}):")
if not potential_results:
print("No relevant trials found for this profile based on the filters and similarity threshold.")
else:
# Prepare formatted output
formatted_output = []
for i, result in enumerate(potential_results):
formatted_output.append(f"\n--- Result {i+1} ---")
formatted_output.append(f"NCT Number: {result['NCT Number']}")
formatted_output.append(f"Study Title: {result['Study Title']}")
formatted_output.append(f"Status: {result['Study Status']}")
formatted_output.append(f"Phases: {result['Phases']}")
formatted_output.append(f"Interventions: {result['Interventions']}")
formatted_output.append(f"Conditions: {result['Conditions']}")
formatted_output.append(f"Brief Summary: {result['Brief Summary']}")
formatted_output.append(f"Primary Outcome: {result['Primary Outcome Measures']}")
formatted_output.append(f"Relevance Score (Semantic Sim): {result['semantic_similarity']:.4f}")
formatted_output.append("-" * 50) # Separator
print("\n".join(formatted_output))
return potential_results # Return the list of result dictionaries
</code>
<code>
# --- Manual Input Section ---
print("\nPlease enter patient information for clinical trial matching.")
# --- Example 1: NSCLC, Stage IV, specific mutations ---
user_cancer_type = "Non-Small Cell Lung Cancer"
user_stage = "Stage 4"
user_biomarkers = "EGFR mutation, PD-L1 positive"
find_relevant_trials(df, drug_embeddings, index_to_embedding_index, model, user_cancer_type, user_stage, user_biomarkers)
# --- Example 2: Breast Cancer, metastatic, HER2-low ---
# user_cancer_type = "Breast Cancer"
# user_stage = "metastatic"
# user_biomarkers = "HER2 low"
# find_relevant_trials(df, drug_embeddings, index_to_embedding_index, model, user_cancer_type, user_stage, user_biomarkers)
# --- Example 3: Prostate cancer, mCRPC, PSMA positive ---
# user_cancer_type = "Prostate cancer"
# user_stage = "metastatic castration-resistant"
# user_biomarkers = "PSMA positive"
# find_relevant_trials(df, drug_embeddings, index_to_embedding_index, model, user_cancer_type, user_stage, user_biomarkers)
# --- Example 4: Urothelial Carcinoma (from your sample data) ---
# user_cancer_type = "Urothelial Carcinoma"
# user_stage = "operable high-risk" # Or try 'bladder cancer' in type
# user_biomarkers = "" # No specific biomarkers mentioned in the sample brief summary
# find_relevant_trials(df, drug_embeddings, index_to_embedding_index, model, user_cancer_type, user_stage, user_biomarkers)
</code>
|
{
"filename": "trials_matching.ipynb",
"repository": "nigat12/ai-treatment-connect",
"query": "transformed_from_existing",
"size": 157773,
"sha": ""
}
|
# main_1.ipynb
Repository: mpnguyen2/dpo
<code>
%reload_ext autoreload
%autoreload 2
</code>
**TRAINING**
#### Helper functions
<code>
from train import train
import time
def train_helper(env_name, num_optimize_iters, warm_up_threshold, zero_order, save_interval):
start_time = time.time()
train(env_name, num_optimize_iters, warm_up_threshold, zero_order, save_interval)
print('Training takes {} hours'.format(int(time.time()-start_time)/3600))
</code>
**Surface modeling**
<code>
# Surface, zero order.
env_name = 'surface'
c = 1.05
num_optimize_iters = [int(10000 * (c**i)) for i in range(21)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=15, zero_order=True, save_interval=5)
</code>
<code>
# Surface, first order.
env_name = 'surface'
c = 1.05
num_optimize_iters = [int(5000 * (c**i)) for i in range(21)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=15, zero_order=False, save_interval=5)
</code>
**Grid-based modeling**
<code>
# Grid, zero order.
env_name = 'grid'
c = 1.05
num_optimize_iters = [int(10000 * (c**i)) for i in range(21)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=15, zero_order=True, save_interval=5)
</code>
<code>
# Grid, first order.
env_name = 'grid'
c = 1.05
num_optimize_iters = [int(5000 * (c**i)) for i in range(21)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=15, zero_order=False, save_interval=5)
</code>
**Molecular dynamics**
<code>
# Molecule zero order.
env_name = 'molecule'
c = 1.5
num_optimize_iters = [int(1000 * (c**i)) for i in range(10)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=5, zero_order=True, save_interval=5)
</code>
<code>
# Molecule first order.
env_name = 'molecule'
c = 1.5
num_optimize_iters = [int(1000 * (c**i)) for i in range(10)]
train_helper(env_name, num_optimize_iters, warm_up_threshold=5, zero_order=False, save_interval=5)
</code>
|
{
"filename": "main_1.ipynb",
"repository": "mpnguyen2/dpo",
"query": "transformed_from_existing",
"size": 17799,
"sha": ""
}
|
# parse_trees_1.ipynb
Repository: eaton-lab/toytree
# Tree Parsing (I/O)
Parsing tree data involves loading a tree topology and associated metadata from a serialized text format into a
data structure. `toytree` loads trees from a variety of text formats (Newick, nexus, NHX) stored in a file, URL, or string, and returns a `ToyTree` class object.
This is made incredibly simple in `toytree` through the general purpose `toytree.tree()` function. In most cases, you can simply call this method on your data (string, file, or URL), without having to even specify the input data type or format.
<code>
import toytree
</code>
<code>
# example newick string
DATA = "((tip1:2,tip2:2):1,tip3:3);"
# load/parse into a ToyTree
tree = toytree.tree(DATA)
tree
</code>
<div class="admonition tip">
<p class="admonition-title">Take Home</p>
<p>
You can parse almost any tree data (file, string, nexus, newick, etc) using <b>toytree.tree()</b>.
</p>
</div>
## Tree data formats
Below are examples of the common Newick, NHX, and Nexus tree data formats. Newick is the base format from which the other two formats are extensions. More details on parsing each format is described further below. While a few additional formats (e.g., JSON or XML) are sometimes used to store tree data, these Newick-based formats are most common.
<code>
# newick: represents a topology using nested parentheses
NEWICK0 = "((,),);"
</code>
<code>
# newick: name strings are usually present for tips as `(label,)`
NEWICK1 = "((tip1,tip2),tip3);"
</code>
<code>
# newick: names can also be present for internal nodes as `()label`
NEWICK2 = "((tip1,tip2)internal1,tip3)internal2;"
</code>
<code>
# newick: edge lengths (dists) are usually present as `()label:dist`
NEWICK3 = "((tip1:2,tip2:2):1,tip3:3);"
</code>
<code>
# newick: support values can be stored in place of internal names `()support`
NEWICK4 = "((tip1,tip2)100,tip3);"
</code>
<code>
# nhx: additional metadata is stored as key=value pairs as `()[meta]`
NHX1 = "((tip1[&trait=2],tip2[&trait=4])[&trait=3],tip3[&trait=1])[&trait=5];"
</code>
<code>
# nexus: newick/NHX data with other code blocks between (begin... end;)
NEXUS1 = """
#NEXUS
begin trees;
translate
1 apple,
2 blueberry,
3 cantaloupe,
4 durian,
;
tree tree0 = [&U] ((1,2),(3,4));
end;
"""
</code>
## Parsing ToyTrees (tldr;)
Parsing tree data is made simple in `toytree` through the general purpose `toytree.tree()` function. For example, this method can parse all of the above data strings correctly without the need of any additional arguments to specify the data or metadata formats. Moreover, it can can parse these data regardless of whether they are entered as a string, or as a file path, or even a public URL. In this way, `toytree.tree()` acts as a sort of swiss army knife for tree data parsing.
<code>
# parse all 7 tree data strings from above into ToyTree objects
data = [NEWICK0, NEWICK1, NEWICK2, NEWICK3, NEWICK4, NHX1, NEXUS1]
trees = [toytree.tree(i) for i in data]
trees
</code>
## Newick format
A `ToyTree` can be flexibly loaded from a range of text formats. When parsing Newick
data it is important to be aware of its limitations. Specifically, that internal node
labels are sometimes used for different purposes, to store either node names, node support
values (as int or floats), and sometimes for other forms of metadata. The `toytree.tree`
function will auto-detect whether these labels should be stored as names or supports based
on their values being numeric or not, however, you can also override this behavior to assign
the values to a feature name of your choice. This is demonstrated below using two examples
of Newick strings with different internal node label types (`NEWICK2` and `NEWICK4`, from above).
### Internal labels as names
If any internal node labels present are non-numeric then they will be parsed and stored as "name" features of Nodes. In the example below the Newick string is parsed into a ToyTree object and its `.get_node_data()` function is called to show the tree's metadata, showing that labels were assigned to 'name'.
<code>
# print newick with str labels for tips and internal nodes
print(f"Newick = {NEWICK2}")
# parse the newick string with .tree()
tree = toytree.tree(NEWICK2)
# show the tree data (labels were assigned to 'name' feature)
tree.get_node_data()
</code>
### Internal labels as support
In contrast to the example above, you can see that the internal labels here are numeric and have thus been stored as "support" features instead of "name", and the internal nodes have names set to the default empty strings. This is the typical format of a Newick string generated by phylogenetic inference software, usually representing some kind of support values. Note that tip nodes/edges do not have support values, nor does the root edge. Support values are actually features of edges, not nodes. This is important for how they are re-oriented when trees are re-rooted (see [Edge Features](/toytree/edge-features)).
<code>
# print newick with str labels for tips and int labels for internal nodes
print(f"Newick = {NEWICK4}")
# parse the newick string with .tree()
tree = toytree.tree(NEWICK4)
# show the tree data (labels assigned to 'support' for internal Node)
tree.get_node_data()
</code>
### Internal labels explicit
As you've seen the use of internal Newick labels can be inconsistent, which is one of the main reasons that the extended Newick format (NHX) was developed, which will be introduced next. Nevertheless, instead of relying on the `toytree.tree` function to automatically parse the internal label as a name or support value, you can optionally enter the feature name you want the values assigned to explicitly using the `internal_labels` arg. For example, you could enter "name", or "support", in which case it will still be parsed as `str` or `float` tyeps, or you can enter any other name to store as a different feature name.
<code>
# parse the newick string with internal str labels and assign
tre0 = toytree.tree(NEWICK2, internal_labels="arbitrary")
# show the tree data where labels were assigned to "arbitrary"
tre0.get_node_data()
</code>
## NHX format
The extended New Hampshire format (NHX) has emerged as a more recent and popular format for tree data storage (although unfortunately the precise rules for the format are not consistently followed). In addition to the standard information in Newick data provided by parentheses (topology) and edge lengths, any additional and arbitrary metadata can be stored within square brackets.
The `toytree.tree()` function will automatically detect if square brackets are present in a Newick string and parse the associated metadata. It is important to note that different programs sometimes vary in the way that they store data inside of the square brackets, and so `toytree.tree` takes a number of additional optional arguments that can be entered to properly parse the NHX metadata. Below are some examples.
Finally, NHX format has the advantage over Newick in that it can distinguish between data that is assigned to Nodes versus Edges in a tree. Data on edges, such as support values, are treated differently than data on nodes, such as trait values, when re-rooting trees (See [Data/Features](/toytree/data) for more on this).
<code>
# only tip Node metadata
NHX1 = "((a[&N=1],b[&N=2]),c[&N=3]);"
# only internal Node metadata
NHX2 = "((a,b)[&N=4],c)[&N=5];"
# both tip and internal Node metadata
NHX3 = "((a[&N=1],b[&N=2])[&N=4],c[&N=3])[&N=5];"
# only edge metadata
NHX4 = "((a:1[&E=1],b:1[&E=2]):1[&E=4],c:1[&E=3]);"
# both node and edge metadata
NHX5 = "((a[&N=1]:1[&E=1],b[&N=2]:1[&E=2])[&N=4]:1[&E=4],c[&N=3]:1[&E=3])[&N=5];"
</code>
<code>
# NHX1 has only tip node data mapped to feature "N"
toytree.tree(NHX1).get_node_data()
</code>
<code>
# NHX5 has all node data mapped to feature "N" and edge data to feature "E"
toytree.tree(NHX5).get_node_data()
</code>
## NEXUS format
The NEXUS format is popular in the field of phylogenetics because it provides a flexible format for storing a variety of information -- both data and instructions -- that can be used by multiple software tools. A NEXUS file starts with a "#NEXUS" header, and then contains one or more *blocks* delimited by "begin" and "end;" statements. For example, a "data" block would start with "begin data" and could contain morphological or molecular data. Another block might include code instructions for the *mrbayes* software, which takes a NEXUS file as input with instructions for an analysis. This could then write results to a "trees" block, which contains one or more Newick or NHX strings. In this way, a NEXUS file can fully describe an analysis from data -> analysis -> trees, as in the example below.
For now, as far as `toytree` is concerned, only the "trees" block is of interest, and all other block are ignored. The `toytree.tree()` function will parse the tree data inside a NEXUS file just as it parses other Newick or NHX strings.
<code>
# nexus: Newick/NHX data with other code blocks between (begin... end;)
NEXUS_EXAMPLE = """
#NEXUS
begin data;
...
end;
begin mrbayes;
...
end;
begin trees;
translate
1 apple,
2 blueberry,
3 cantaloupe,
4 durian,
;
tree tree0 = [&U] ((1,2),(3,4));
end;
"""
</code>
<code>
# parse NEXUS file and show tree data
tree = toytree.tree(NEXUS_EXAMPLE)
tree.get_node_data()
</code>
## Parsing MultiTrees
Sometimes data from multiple trees are stored together in a single file, such as the results of a bootstrap analysis, or a posterior distribution of trees from a Bayesian phylogenetic inference. `toytree` can parse and load all trees in a multiple tree input using the `toytree.mtree` function. This returns a `MultiTree` object (see [MultiTree](/toytree/multitree)), which has methods that can apply to sets of trees, and from which individual `ToyTrees` can be indexed and extracted.
<code>
# a str with Newick data separated by new lines
MULTILINE_NEWICK = """
(((a:1,b:1):1,(d:1.5,e:1.5):0.5):1,c:3);
(((a:1,d:1):1,(b:1,e:1):1):1,c:3);
(((a:1.5,b:1.5):1,(d:1,e:1):1.5):1,c:3.5);
(((a:1.25,b:1.25):0.75,(d:1,e:1):1):1,c:3);
(((a:1,b:1):1,(d:1.5,e:1.5):0.5):1,c:3);
(((b:1,a:1):1,(d:1.5,e:1.5):0.5):2,c:4);
(((a:1.5,b:1.5):0.5,(d:1,e:1):1):1,c:3);
(((b:1.5,d:1.5):0.5,(a:1,e:1):1):1,c:3);
"""
# parse with .mtree
mtree = toytree.mtree(MULTILINE_NEWICK)
mtree
</code>
<code>
# a Nexus str with trees in a trees block
MULTI_N5XUS = """
#NEXUS
begin trees;
translate
1 a,
2 b,
3 c,
4 d,
5 e,
;
tree 1 = [&R] (((1:1,2:1):1,(4:1.5,5:1.5):0.5):1,3:3);
tree 2 = [&R] (((1:1,4:1):1,(2:1,5:1):1):1,3:3);
tree 3 = [&R] (((1:1.5,2:1.5):1,(4:1,5:1):1.5):1,3:3.5);
tree 4 = [&R] (((1:1.25,2:1.25):0.75,(4:1,5:1):1):1,3:3);
tree 5 = [&R] (((1:1,2:1):1,(4:1.5,5:1.5):0.5):1,3:3);
tree 6 = [&R] (((2:1,1:1):1,(4:1.5,5:1.5):0.5):2,3:4);
tree 7 = [&R] (((1:1.5,2:1.5):0.5,(4:1,5:1):1):1,3:3);
tree 8 = [&R] (((2:1.5,4:1.5):0.5,(1:1,5:1):1):1,3:3);
end;
"""
# pars5 with .mtree
mtree = toytree.mtree(MULTI_N5XUS)
mtree
</code>
If you call `toytree.mtree` on a file containing a single tree then it will simply return a `MultiTree` object containing only a single `ToyTree` within it. If you call `toytree.tree` on a file containing multiple trees it will return the first tree in the file as a `ToyTree`, but will also print a warning to make sure you know that the input contained multiple trees.
<code>
# calling .mtree on a single tree input is OK
toytree.mtree(NEWICK1)
</code>
<code>
# calling .tree on a multiple tree input is also OK, but raises a WARNING
toytree.tree(MULTILINE_NEWICK)
</code>
### Loading trees from URLs
A convenient feature of `toytree.tree` is the ability to laod tree data from a public URI. If you provide a string as input that begins with "http" then the str data of that URI will be checked for valid tree data. If so, it is returned as a tree. You can thus store your trees on any public site, such as a GitHub repo, and easily load it in without having to write out a long file path.
<code>
toytree.tree("https://eaton-lab.org/data/Cyathophora.tre")
</code>
|
{
"filename": "parse_trees_1.ipynb",
"repository": "eaton-lab/toytree",
"query": "transformed_from_existing",
"size": 42776,
"sha": ""
}
|
# Spatial_Transcriptomics_1.ipynb
Repository: nunososorio/SingleCellGenomics2024
[](https://colab.research.google.com/github/nunososorio/SingleCellGenomics2024/blob/main/5_Friday_April12th/Spatial_Transcriptomics.ipynb)
<img src="https://github.com/nunososorio/SingleCellGenomics2024/blob/main/logo.png?raw=true" alt="AnnData" style="width:600px; height:auto;"/>
# Spatial transcriptomics
In this part of the course we will focus on spatial transcriptomics data. Spatial transcriptomic technologies allow for the systematic measurement of gene expression levels throughout the tissue sample, increasing our understanding of cellular organisations and interactions within tissues while also providing biological insights in a wide range of subject and diseases. Multiple types of spatial techniques have been developed and they vary within spatial resolution, multiplexing capabilities, sensitivity, coverage, and throughput.
The different developed methods can by divided into four categories:<br>
- **Sequencing-based:** 10X Genomics Visium, Stereo-seq, Slide-seq, Light-seq;<br>
- **Probe-based:** NonoString GeoMx;<br>
- **Imaging-based:** NanoString CosMx SMI, STARmap, MERFISH, seqFISH; <br>
- **Image-guided spatially resolved:** NICHE-seq, Geo-seq, Zip-seq.<br>
Some methodologies also allow to access different types of omics in addition to transcriptomics (RNA), such as DNA, protein, metabolite, chromatin accessibility, histone modification , among others. For more information on this topic you can check the following paper doi: 10.3390/cells12162042
# Load packages
In this tutorial we'll be using the following packages in order to perform the basis of the spatial transcriptomics data analysis.
*Scanpy* will be used to perform the data analysis and visualization, as donne on the previous notebook for scRNA-seq.
*Pandas* and *Numpy* packages will be used to perform data matrices manipulation.
*seaborn* and *matplotlib* will be used in some cases to plot the results.
For more advanced analysis we can check on *squidpy*.
<code>
! pip install scanpy > _
</code>
<code>
import scanpy as sc
import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
sc.settings.verbosity = 3
</code>
# Data set
This dataset was retrieved form the GEO database using the accession number ***GSE226208*** and the paper is entitled "*Shared inflammatory glial cell signature after brain injury, revealed by spatial, temporal and cell-type-specific profiling of the murine cerebral cortex*" and has the following doi: https://doi.org/10.1101/2023.02.24.529840. The goal of this paper was to understand the mechanisms working after traumatic brain injury (TBI). This study is composed by two dataset: one of spatial data, which the one we are using and one of scRNA-seq data, which we'll not use in this course. These tow were used to analyse the transcriptomic signature of the mouse injured cerebral cortex.
The main results were the identification of specific states for microglia, astrocytes, and OPCs which comprised some genes related to injury, including genes related to inflammatory responses of the innate immune system (Cxcr3).
The spatial dataset is composed by 2 samples, one from healthy mouse brain (Intact/ctl) and another from a 3 days post-injury brain (3DPI/dpi3). The brain injuries were stab-wonds induced on the cerebral cortex, affecting only the grey matter. Each spatial transcriptomics slide contains 2 tissue slices from each condition, one with the normal orientation and the other one upside-down.
As as summary, this study serves as a spatial and temporal map of cell-type specific mouse cortex, revealing the inflammatory signatures of glial cells after trauma.
The technique used to sequence this samples was the Visium technology from 10X, where each slice contains 5000 spots of 55$\mu with unique barcodes, and each spot can capture the transcripts from 1 to 10 cells, so the number of cells per spot will depend on the size of each cell and its neighbours. To capture the mRNA, the tissue needs to be permeabilized and then the mRNA will bind to the capture oligonucleotides present in each spot. Then the mRNA is synthetized into cDNA and the sequencing library is prepared. To get the spatial information before permeabilizing the tissue we need to stain the tissue with H&E or IF and do the imaging.
Here we start already with the data matrix instead of the FASTQ files, which means that the authors already ran the SpaceRanger, which is the equivalent of CellRanger but for spatial data.
Here is a figure to explain more about the dataset we will use:
<img src="https://github.com/nunososorio/SingleCellGenomics2024/blob/main/5_Friday_April12th/overview_figure.png?raw=true" alt="AnnData" style="width:600px; height:auto;"/>
# Download the data to use in this tutorial
<code>
! wget https://github.com/nunososorio/SingleCellGenomics2024/raw/main/5_Friday_April12th/Data.zip
! unzip Data.zip
</code>
# Load the data
<code>
ctl = sc.read_visium(path='Data/GSE226208/intact')
dpi3 = sc.read_visium(path='Data/GSE226208/3dpi/')
</code>
# Pre-processing
## Check data structure
### Exercise
Check the data structure for each condition
<code>
#write your code here
</code>
<code>
#write your code here
</code>
In the following steps we'll make the *var_names* (gene names) unique for each data set, create a raw data object, where the raw data of each sample will be saved in case we need the raw counts matrix on the downstream analysis.
Lastly, we'll create a new column on the *obs* slot, named *Condition*, containing samples' origin information, wether the data is from the Intact cortex or 3DPI.
<code>
ctl.var_names_make_unique()
dpi3.var_names_make_unique()
</code>
<code>
ctl.raw = ctl.copy()
dpi3.raw = dpi3.copy()
</code>
<code>
ctl.obs['Condition'] = 'Intact'
dpi3.obs['Condition'] = '3DPI'
</code>
Here we are setting colors to each condition, being the Intact sample pastel blue and the 3DPI sample pastel red. this helps when plotting since we know already each colors represents each condition.
<code>
ctl_col = '#a1c9f4'
dpi3_col = '#ff9f9b'
</code>
Next we calculate some basic metrics for each sample, so then we can compare them and make decisions on the next steps to be executed. The following command will calculate some quality metrics, being the most important and the one that we'll use the total number of counts and the number of genes by counts.
<code>
ctl_m = ctl.copy()
dpi3_m = dpi3.copy()
</code>
<code>
sc.pp.calculate_qc_metrics(ctl_m, inplace=True)
sc.pp.calculate_qc_metrics(dpi3_m, inplace=True)
</code>
<code>
print(f"{ctl_m} \n\n {dpi3_m}")
</code>
Here the 2 metrics mentioned above wil be plotted side by side for each sample and the plots will be colored according to the chosen colors.
<code>
fig, axs = plt.subplots(1,4, figsize=(15,4))
fig.suptitle('Covariates for filtering')
sns.histplot(ctl_m.obs['total_counts'], kde=False, bins=60, ax = axs[0], color=ctl_col)
sns.histplot(dpi3_m.obs['total_counts'], kde=False, bins=60, ax = axs[1], color=dpi3_col)
sns.histplot(ctl_m.obs['n_genes_by_counts'], kde=False, bins=60, ax = axs[2], color=ctl_col)
sns.histplot(dpi3_m.obs['n_genes_by_counts'], kde=False, bins=60, ax = axs[3], color=dpi3_col)
</code>
**NOTE:** Maybe here we can ask how they perceive the samples, if their quality is similar or not, based on the distribution of the histograms since we will use it to decide if we merge the samples or not.
# Merge data
When the project we are working on is composed by more than one dataset from different samples, we can merge all the small datasets into a major one, containing all the samples. This helps on the filtering process since we'll be working with only one big object instead of several small ones. Additionally this makes the filtering process more uniform among samples. However, when we see that the quality of the samples is quite different between then, the filtering should be perform in each sample individually and inly after that we should merge the different samples.
## Merge counts data
Since we are working with spatial data, to merge our samples into one object we need to perform some steps before in order to allow us to visualize the data projected on the image. For that we need to use the package *stlearn*. Tis package also offers a wide range of possible downstream analysis and is worth to check if you need analyze spatial data in the future.
To use this package we only need to install it as follows on the next code cell.
<code>
! pip install stlearn > _
</code>
Once installed we need to load it, and then convert our individual dataset from scanpy format to the stlearn format. This step will add two new columns to the *obs* slot called *imagecol* and *imagerow*, and these are the ones used to merged both images. The code used to perform this step used also the *numpy* package to manipulate the array containing the image information/coordinates.
<code>
import stlearn as st
</code>
<code>
ctl = st.convert_scanpy(ctl)
dpi3 = st.convert_scanpy(dpi3)
</code>
Once our data is converted we can now merge both samples into one big dataset.
To merge two or more samples into one data object we use th scanpy function *concatenate* as follows. We select one of the samples to be the one where all the rest of the samples will be concatenated to. In our case we'll concatenate the 3DPI sample to the Intact sample.
**NOTE:** Here we can ask if they think we should merge both samples into one object.
<code>
adata = ctl.concatenate(dpi3)
</code>
### Exercise
Check the data structure and also check both *var* and *obs* slots of the data. Do you see any difference when comparing to scRNA-seq data?
<code>
#write your code here
</code>
<code>
#write your code here
</code>
<code>
#write your code here
</code>
## Merge images data
Even though *scanpy* does not offer an option to integrate spatial images, we can do it manually. However, we first need to convert our samples from the *scanpy* format to *stlearn* format as we already did when merging the data counts. If we perform this step we do not need to split our data into two distinct datasets to perform visualization on the spatial images.
To merged our images we will also use the package *numpy* since we need to manipulate the arrays containing the coordinates of each image. This stepp will allow to plot each slice side by slice as it was only one image, avoid the need to plot each sample separately.
<code>
# Initialize the spatial
adata.uns["spatial"] = ctl.uns["spatial"]
# Horizontally stack 2 images from section 1 and section 2 datasets
combined = np.hstack([ctl.uns["spatial"]["D_Intact"]["images"]["hires"],
dpi3.uns["spatial"]["A_3dpi_V"]["images"]["hires"]])
# Map the image to the concatnated adata object
adata.uns["spatial"]["D_Intact"]["images"]["hires"] = combined
# Manually change the coordinate of spots to the right
adata.obs.loc[adata.obs.batch == "1","imagecol"] = adata.obs.loc[adata.obs.batch == "1","imagecol"].values + 2000
# Change to the .obsm["spatial"]
factor = adata.uns["spatial"]["D_Intact"]["scalefactors"]["tissue_hires_scalef"]
adata.obsm["spatial"] = adata.obs[["imagecol","imagerow"]].values / factor
</code>
### Exercise
Save the merged data object into your working directory.
<code>
# Write you code here
</code>
# QC
One of the first steps when preparing the data to perform quality control e to get QC metrics such as the one calculated previously (total counts and number of genes by counts). Here, additionally we will also get mitochondrial (mt) and ribossomal (ribo) genes and then calculate the percentage of mitochondrial and ribossomal counts. To get which genes are mitochondrial or ribossomal genes we we add a new column to the *var* slot with True or False values, classifying the genes as mt or ribo. For taht we use the function *startswith* to get the genes that start with ***mt*** or by ***Rps***/***Rpl***, corresponding to mitochondrial and ribossomal genes, accordingly.
For that we will use the function previously used *sc.pp.calculate_qc_metrics*
## QC metrics
<code>
adata.var["mt"] = adata.var_names.str.startswith("mt-")
adata.var["ribo"] = adata.var_names.str.startswith(("Rps","Rpl"))
#fill in the blanks
#Calculate qc metrics for mt and ribo genes
sc.pp.calculate_qc_metrics(, qc_vars=[], inplace=True)
</code>
<code>
adata
</code>
## Cell cycle
As for the scRNA-seq data analysis, here we can also add cell cycle information to the data.
### Exercise
Search for the files containing the cell cycle information inside the Data folder you downloaded at the begining of the notebook and load it on the following cell of code. Then calculate the cell cycle score.
<code>
#Fill in the blank spaces
#Load the files containing the genes to each phase of the cell cycle
s_genes = [x.strip() for x in open( )]
g2m_genes = [x.strip() for x in open( )]
</code>
<code>
#fill in the blank spaces
sc.tl.score_genes_cell_cycle( )
</code>
Then we plot the calculated values and we chose the thresholds to our data. This visualization can be performed by using histograms or violin plots.
<code>
fig, axs = plt.subplots(1,4, figsize=(15,4))
fig.suptitle('Covariates for filtering')
sns.histplot(adata.obs['total_counts'], kde=False, ax = axs[0])
sns.histplot(adata.obs['n_genes_by_counts'], kde=False, bins=60, ax = axs[1])
sns.histplot(adata.obs['pct_counts_mt'], kde=False, bins=60, ax = axs[2])
sns.histplot(adata.obs['pct_counts_ribo'], kde=False, bins=60, ax = axs[3])
</code>
Here we are doing a zoom in on the histogram to choose the lowe threshold for each filtering parameter.
<code>
fig, axs = plt.subplots(1, 4, figsize=(15, 4))
sns.histplot(adata.obs["total_counts"][adata.obs["total_counts"] < 10000], kde=False, bins=60, ax=axs[0])
sns.histplot(adata.obs["n_genes_by_counts"][adata.obs["n_genes_by_counts"] < 4000], kde=False, bins=60, ax=axs[1])
sns.histplot(adata.obs["pct_counts_mt"][adata.obs["pct_counts_mt"] > 25], kde=False, bins=60, ax=axs[2])
sns.histplot(adata.obs["pct_counts_ribo"][adata.obs["pct_counts_ribo"] > 10], kde=False, bins=60, ax=axs[3])
</code>
<code>
sc.pl.violin(adata, keys=['total_counts','n_genes_by_counts', 'pct_counts_mt', 'pct_counts_ribo'], rotation=90, multi_panel=True)
</code>
### Exercise
Chose the thresholds to filter the data. Since the resolution is not single cell, we should take it into account when choosing the values to filter the data. here we are not filtering out cells but spots that may contain more than one cell, keep that in mind.
**Hint**: The code used to filter spatial data is the same used to filter the scRNA-seq data.
<code>
#write your code here
#filter counts
sc.pp.filter_cells(adata, min_counts =)
sc.pp.filter_cells(adata, max_counts =)
#filter genes
sc.pp.filter_cells(adata, min_genes =)
sc.pp.filter_genes(adata, min_cells =)
#filter mito genes
#write your code here
#filter ribo genes
#write your code here
</code>
<code>
sc.pl.violin(adata, keys=['total_counts','n_genes_by_counts', 'pct_counts_mt', 'pct_counts_ribo'], rotation=90, multi_panel=True)
</code>
### Exercise
Save the filtered data into your working directory
<code>
# Write you code here
</code>
# Normalization and logaritmization
After filtering the cells with bad quality we proceed to data normalization and logaritmization. Here we will use the default method to normalize the data, which normalizes each cell by total counts over all genes. This makes that every cell will have the same total count after normalization. However other methods could have been used as SCTransform or GLM-PCA, whose have an higher sensitivity for normalization.
<code>
sc.pp.normalize_total(adata, inplace = True)
sc.pp.log1p(adata)
</code>
In this case we will not perform regression of any variable, such as percentage of mt genes or cell cycle since this variables may play a role on the variation of the cell types after injury.
# Dimentionality reduction
Once filtering is done the next step, as in scRNA-seq data, is to perform a reduction of the dimension of the dataset we are working with.
The first step will be to select the high variable genes for our data, followed by principal component analysis and uniform manifold approximation and projection.
## Highly Variable Genes (HVG)
In this case we will decide how many genes we want to keep, which is 4000. This value is up to you, many tutorials usually uses 2000 genes, but for the purpose of this course we will keep a little more genes.
There is also the possibility to define different thresholds to different metrics and the function will select the genes that passes those thresholds as the HGV of our dataset. The used method will depend on the data your working with and om which one you think fits the best your goal on the analysis. For more information you can check the documentation of the following function *highly_variable_genes*.
### Exercise
Calculate the HVG selectin the top 4000 genes.
<code>
#fill in the blanks
sc.pp.highly_variable_genes(, inplace=True)
</code>
<code>
sc.pl.highly_variable_genes(adata)
</code>
Here we keep only the HVG on the data, removing all the genes that are not highly variable
<code>
var_genes_all = adata.var.highly_variable
adata = adata[:, var_genes_all]
adata
</code>
## Principal Component Analysis (PCA)
The PCA is performed with the same goal as on the analysis of scRNA-seq data, which is to reduce the dimension of the dataset, reduction it to the lower number possible of principal components (PCs) that allow to retain the maximum variation within the data.
Here the PCA will be performed with the default settings, nonetheless these values can be adjusted to the data we are working with.
In our case we don't need to set the parameter ***use_highly_variable*** since our dataset only has the HVG already.
### Exercise
Calculate the PCA using the function *sc.pp.pca()*
<code>
#write here your code
</code>
Next we plot the variance of the calculated PCs in order to select how many we want to keep. Usually the rule is that we select the last one before the curve of the plot starts to flatten. However we can select a little fewer or a little more.
### Exercise
Plot the variance ratio for the calculated PCs, and show 50 PCs on the plot.
**Hint:** The code is the same as the one used for scRNA-seq data.
<code>
#write here your code
sc.pl.pca_variance_ratio()
</code>
## Uniform Manifold Approximation and Projection (UMAP)
This method is used to visualize the data in a 2D way, making it simpler. To compute the UMAP we use the number of PC we selected on the previous step.
### Exercise
Compute the UMAP using the number of PC that you think fits the best. Use the functions *sc.pp.neighbors()* and *sc.tl.umap()*.
<code>
#fill in the blanks and write your code
sc.pp.neighbors(n_neighbors=15)
</code>
Now we can perform data visualization in a reduces dimension, such as visualize the metrics calculated on the CQ step in a 2D space.
### Exercise
Plot the metrics used on quality control and the variable *Condition* on the UMAP, so you can see where are the spots with the higher and lower values to each of those metrics.
**Metrics:** *'total_counts'*, *'n_genes_by_counts'*, *'pct_counts_mt'*, *'pct_counts_ribo'*.
<code>
#fill in the missing values from the function
sc.pl.umap(adata, color=, ncols=2, cmap='viridis')
sc.pl.umap(adata, color=, palette=[dpi3_col, ctl_col])
</code>
# Clustering
For spatial data, clustering can be performed as for scRNA-seq data, and the same principals apply here.
### Exercise
Fill in the spaces with commented line with your code. To make it easier we will cerate a list where we will save the names we gave to each cluster to use it further on the analysis.
<code>
#create here you empty list
# complete the commented lines
for in [0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 1.1, 1.2, 1.3, 1.4]: #chose the name of you iteration variable so in each loop it will assume a different resolution value
sc.tl.leiden(, resolution= , key_added='clusters_%s'% )
#append the name of each clsuter resolution calculated to the list you created use the following string formating to name you cluster resolution eg. clusterin with resolution 0.1 will be called clusters_0.1
('clusters_%s'% )
</code>
### Exercise
Plot all the cluster resolution on a UMAP, use the list we created above to make it easier.
<code>
# fill in the wite spaces
sc.pl.umap( ncols=3, wspace = 0.6)
</code>
Since scanpy does not offer a tool to integrate the images of the dataset, now we need to split our dataset again into two different objects, but now the all the pre processing and dimensionality reduction of the data is already performed. For that we split the our ***adata*** object into ***adata_ctl*** and ***adata_dpi3*** using the column ***Condition*** on the *obs* slot of the data, by indicating the different conditions to split.
<code>
adata_ctl = adata[adata.obs['Condition'] == 'Intact',:]
adata_dpi3 = adata[adata.obs['Condition'] == '3DPI',:]
</code>
<code>
adata
</code>
<code>
print(f"{adata_ctl} \n\n {adata_dpi3}")
</code>
Once our data is split in two we can now plot our data into the spatial images
### Exercise
Plot the different control metrics for each condition on the spatial image: *'total_counts'*, *'n_genes_by_counts'*, *'pct_counts_mt'*, *'pct_counts_ribo'*.
<code>
sc.settings.set_figure_params(dpi=150)
#fill in the empty spaces
sc.pl.spatial(, img_key = "hires", color=)
sc.pl.spatial(, img_key = "hires", color=)
</code>
### Exercise
Now plot the clusters on the spatial image for each condition. Complete the lines with missing code.
**Hint:** remember that we created a list with all cluster names previously!
<code>
#write your code here
sc.pl.spatial(, img_key = "hires", color=, size=1.5, wspace=0.5)
sc.pl.spatial(, img_key = "hires", color=, size=1.5, wspace=0.5)
</code>
### Exercise
Now we will plot the clusters on the integrated image of the data set (Both intact and 3DPI samples side by side). This will help to note differences on the clusters between samples.
To do this exercise we weill use a for loop to iterate the cluster names and the function *st.pl.cluster_plot()* from *stleran* to plot the clusters on the integrated image. Fill in the empty spaces.
<code>
# fill the blanks
for in :
st.pl.cluster_plot(a, use_label=, crop=False, size=1.4, cell_alpha=1)
</code>
<code>
adata
</code>
# Rank marker genes
Know we rank the genes in each cluster the get it's marker genes to further annotated them with the according cell type.
### Exercise
Using a for loop to iterate the names fo the clusters, the *sc.tl.rank_genes_groups()* to rank the genes and the function *sc.pl.rank_genes_groups_dotplot()* to plot the marker genes using a dotplot, rank and plot the marker genes for each cluster. The method to rank the marker genes will be the *wilcoxon* method.
Fill in the blank spaces.
<code>
#complete the commented lines
for in :
#here print the name of the resolution beiing used in each iteration
sc.tl.rank_genes_groups(, , inplace = True, , use_raw=False)
sc.pl.rank_genes_groups_dotplot(, n_genes=15, groupby = , vmax=5)
</code>
<code>
adata
</code>
# Cluster annotation
When annotating the different clusters from a spatial transcriptomics dataset we need to keep in mind that each spot may contain more than one cell, depending on the size of the cell in that spot. For example, immune cells are much smaller than neurons or oligodendrocytes. For that reason some times it can be difficult to attribute only one cell type to a cluster, in that case we can use a more general classification.
**Question:** Do you thin k that we may have any cluster that may be composed by different cell types? To help you chose the best resolution you can go and check on the paper how many clusters they considered.
**NOTE:** To annotate your clusters without a reference data set you can use the following mouse brain expression map (http://mousebrain.org/adolescent/genesearch.html) to check which cells express the genes obtained to each cluster. Also in case you need to check where a certain region of the brain is located you can check this atlas https://atlas.brain-map.org/.
### Exercise:
Annotated the cell types you identified, first by cell types and then by cells groups. This means that first you consider all cell types and then the different subtypes of cells will be grouped together. For tat you will create two new columns on the *obs* slot of the adata object called **Cell_type** and **Cell_type_groups**.
<code>
#Fill in the blanks
adata.obs[] = adata.obs[].replace({ })#here write a dict where you keys are the numbers of your cluster on your chosen resolution and the values are the corresponding cell type
</code>
<code>
#write your code here
#Now create the obs Cell_type_groups, using the same cluster resolution used above, but group by cell groups, avoiding subtypes.
</code>
### Exercise
Plot the cell types into the integrated spatial images, so you can see where the clusters are. Use the function already previously used *st.pl.cluster_plot()*
<code>
#fil in the the missing values
st.pl.cluster_plot(, use_label=, crop=False, size=1.4, cell_alpha=1)
st.pl.cluster_plot(, use_label=, crop=False, size=1.4, cell_alpha=1)
</code>
### Exercise
Plot both cell type annotations and Condition using the UMAP projection.
<code>
#write your code here
</code>
### Exercise
Calculate the marker genes as done previously for the different cluster resolutions, but now use the **Cell_type** annotation, and plot them using a dotplot. For that use the following functions: *sc.tl.rank_genes_groups()*, *sc.pl.rank_genes_groups_dotplot()*.
<code>
#Fill in the blank spaces
sc.tl.rank_genes_groups(, , inplace = True, key_added="wilcoxon_Cell_type", method='wilcoxon', use_raw=False)
sc.pl.rank_genes_groups_dotplot(, n_genes=15, key="wilcoxon_Cell_type", groupby = 'Cell_type', vmax=5)
</code>
**NOTE:** As you can see the it can be quite difficult to find the most specific genes for each cluster. In cases like this we can add some filtering parameters to our rank genes function that will clean a little bit the genes selected to caracterize the cluster. This step can also be done before cluster annotation if needed.
### Exercise
Using the function *sc.tl.filter_rank_genes_groups()* and the following filtering parameters filter the rank genes and plot them using the function *sc.pl.rank_genes_groups_dotplot()*
**Parameters:** *min_in_group_fraction=0.1*, *max_out_group_fraction=0.5*, *min_fold_change=0.25*.
Fill in the blank sapces.
<code>
#fill in the spaces
sc.tl.filter_rank_genes_groups(, , key = "wilcoxon_Cell_type", key_added='wilcoxon_filtered_leiden_Cell_type' )
#fill in the spaces to plot the dotplot
sc.pl.rank_genes_groups_dotplot(, n_genes=15, key="wilcoxon_filtered_leiden_Cell_type" , groupby="Cell_type", vmin=0, vmax=5)
</code>
### Exercise
Perform the same two exercises you did for ranking the *Cell_type* marker genes, but now do it with the **Cell_type_groups**.
**Hint:** Don't forget to change the name of the function variables.
<code>
#write your code here
</code>
<code>
#write your code here (filter the range genes for cell_type_groups)
</code>
# Marker genes
### Exercise
Based on the dot plot above select 3 marker genes that you consider the best ones to represent the cluster. In case you know some canonical markers for the cell types identified you can also use those ones in addition to the ones on the dotplot. Do this step for both annotations (*Cell_type* and *Cell_type_groups*) and plot then on the spatial map, on the UMAP, and if you want you can also use a dot plot and group it by clusters with their marker genes.
<code>
#create your list here
</code>
### Cell type
#### Spatial image
<code>
st.settings.set_figure_params(dpi=120)
#fill in the blanks to plot each gene individually
for in :
st.pl.gene_plot(, gene_symbols=, use_raw=True, size=0.5, cmap='viridis')
</code>
#### UMAP
<code>
#fill the blanks
sc.pl.umap(, , use_raw=False, ncols=3, cmap='viridis', vmax=5)
</code>
#### Dot plot
To plot the marker genes using a dot plot you need to create a dictionary, where the keys are the different cell types, and the values are a list with the 3 marker genes you selected to each cell type.
<code>
#create your dictionary here
</code>
<code>
#Fill the blank on the code
dp = sc.pl.dotplot(, , , return_fig=True, vmax=5, cmap='Reds')
dp.add_totals().show() #this line of code is just to add a barplot at the end of the dot plot to show the number of cells inside each cluster
</code>
### Cell type groups
<code>
#create here your list with the 3 marker genes you selected to each cell group
</code>
#### Spatial image
<code>
#write your code here
st.settings.set_figure_params(dpi=120)
</code>
#### UMAP
<code>
#write your code here
</code>
#### Dot plot
<code>
#create here you dictionary
</code>
<code>
#Fill the blank on the code
</code>
# Final remarks
In this tutorial we focus on the most basic steps to analyse spatial data, starting from a counts matrix. If you want to take this analisis on step forward you can look into *stlearn* package (https://stlearn.readthedocs.io/en/latest/index.html), where you can find more downstream analysis such as spatial trajectory, cell-cell interation, cell-type deconvolution, and other types of analysis and different plotting.
As a next step it would be inte resting to perform cell type deconvolution, to really have an idea of the cell types present in each spot. Another nice option would be to an annotated dataset with the same type of experiment and transfer the cell annotations to the spatial data.
It is also possible to combine spatial data with scRNA-seq data. This is usually done to infer where certain genes identified on the scRNA-seq data are expressed on the spatial map.
|
{
"filename": "Spatial_Transcriptomics_1.ipynb",
"repository": "nunososorio/SingleCellGenomics2024",
"query": "transformed_from_existing",
"size": 52807,
"sha": ""
}
|
# LDA.ipynb
Repository: ankitvgupta/rnaseqtopicmodeling
|
{
"filename": "LDA.ipynb",
"repository": "ankitvgupta/rnaseqtopicmodeling",
"query": "transformed_from_existing",
"size": 13766,
"sha": ""
}
|
# demo1_2.ipynb
Repository: ZJUFanLab/bulk2space
## Demonstration of Bulk2Space on demo1 dataset
### Import Bulk2Space
<code>
from bulk2space import Bulk2Space
model = Bulk2Space()
</code>
### Decompose bulk-seq data into scRNA-seq data
Train β-VAE model to generate scRNA-seq data
<code>
generate_sc_meta, generate_sc_data = model.train_vae_and_generate(
input_bulk_path='tutorial/data/example_data/demo1/demo1_bulk.csv',
input_sc_data_path='tutorial/data/example_data/demo1/demo1_sc_data.csv',
input_sc_meta_path='tutorial/data/example_data/demo1/demo1_sc_meta.csv',
input_st_data_path='tutorial/data/example_data/demo1/demo1_st_data.csv',
input_st_meta_path='tutorial/data/example_data/demo1/demo1_st_meta.csv',
ratio_num=1,
top_marker_num=500,
gpu=0,
batch_size=512,
learning_rate=1e-4,
hidden_size=256,
epoch_num=20,
vae_save_dir='tutorial/data/example_data/demo1/predata/save_model',
vae_save_name='demo1_vae',
generate_save_dir='tutorial/data/example_data/demo1/predata/output',
generate_save_name='demo1')
</code>
<code>
generate_sc_meta
</code>
<code>
generate_sc_data
</code>
Load trained β-VAE model to generate scRNA-seq data
<code>
generate_sc_meta, generate_sc_data = model.load_vae_and_generate(
input_bulk_path='tutorial/data/example_data/demo1/demo1_bulk.csv',
input_sc_data_path='tutorial/data/example_data/demo1/demo1_sc_data.csv',
input_sc_meta_path='tutorial/data/example_data/demo1/demo1_sc_meta.csv',
input_st_data_path='tutorial/data/example_data/demo1/demo1_st_data.csv',
input_st_meta_path='tutorial/data/example_data/demo1/demo1_st_meta.csv',
vae_load_dir='tutorial/data/example_data/demo1/predata/save_model/demo1_vae.pth',
generate_save_dir='tutorial/data/example_data/demo1/predata/output',
generate_save_name='demo1_new',
ratio_num=1,
top_marker_num=500)
</code>
### Decompose spatial barcoding-based spatial transcriptomics data into spatially resolved single-cell transcriptomics data
Train deep-forest model to generate spatially resolved single-cell transcriptomics data
<code>
df_meta, df_data = model.train_df_and_spatial_deconvolution(
generate_sc_meta,
generate_sc_data,
input_st_data_path='tutorial/data/example_data/demo1/demo1_st_data.csv',
input_st_meta_path='tutorial/data/example_data/demo1/demo1_st_meta.csv',
spot_num=500,
cell_num=10,
df_save_dir='tutorial/data/example_data/demo1/predata/save_model/',
df_save_name='deom1_df',
map_save_dir='tutorial/data/example_data/demo1/result',
map_save_name='demo1',
top_marker_num=500,
marker_used=True,
k=10)
</code>
<code>
df_meta
</code>
<code>
df_data
</code>
Load trained deep-forest model to generate spatially resolved single-cell transcriptomics data
<code>
df_meta, df_data = model.load_df_and_spatial_deconvolution(
generate_sc_meta,
generate_sc_data,
input_st_data_path='tutorial/data/example_data/demo1/demo1_st_data.csv',
input_st_meta_path='tutorial/data/example_data/demo1/demo1_st_meta.csv',
spot_num=500,
cell_num=10,
df_load_dir='tutorial/data/example_data/demo1/predata/save_model/deom1_df',
map_save_dir='tutorial/data/example_data/demo1/result', # file_dir
map_save_name='demo1_new', # file_name
top_marker_num=500,
marker_used=True,
k=10)
</code>
|
{
"filename": "demo1_2.ipynb",
"repository": "ZJUFanLab/bulk2space",
"query": "transformed_from_existing",
"size": 56277,
"sha": ""
}
|
# scRNAseq_Analysis_PartI_sample8.ipynb
Repository: SchoberLab/YF
# Analysis Part I - Preprocessing Sample 8
<code>
%load_ext autoreload
</code>
<code>
%matplotlib inline
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
warnings.filterwarnings(action='ignore')
</code>
<code>
import os
import scanpy as sc
import scirpy as ir
import anndata as ann
import numpy as np
import pandas as pd
import seaborn as sb
import matplotlib.pyplot as plt
from matplotlib import rcParams
from mudata import MuData
import mudata
import tarfile
import warnings
from glob import glob
import muon as mu
</code>
<code>
%autoreload 2
import sys
sys.path.append('..')
import utility.annotation as utils_annotation
import utility.representation as utils_representation
import utility.visualisation as utils_vis
</code>
<code>
sc.settings.set_figure_params(dpi=150)
sc.settings.verbosity = 3
sc.set_figure_params(vector_friendly=True, color_map='viridis', transparent=True)
sb.set_style('whitegrid')
</code>
Samples:
- Sample 4:
- B19_d14 #HA1
- B19_d365 #HA2
- B40_d14 #HA3
- B40_d365 #HA4
- B46_d14
- B46_d21
- B27_d21 #reality d22
- B41_d90
Dextrameres:
- NS4B214-222 -- LLWNGPMAV (A*02:01) -- TTGGCGATTCCTCCA
<code>
#Define the lists for later
hashtags = [f'sample{i}' for i in range(1, 9)]
epitope_ids = ['NS4B214']
cite_seqs = ['CD45RA', 'CCR7-1', 'CD95', 'CD62L']
feature_barcode_ids = hashtags + epitope_ids + cite_seqs
</code>
<code>
##Read data
# GEX data
datafile = "/media/agschober/HDD12/3_scRNA-Seq_Sina/Cellranger_output/2nd_Experiment/run4/outs/per_sample_outs/run4/count/sample_filtered_feature_bc_matrix.h5"
adata = sc.read_10x_h5(datafile, gex_only=False)
adata.var_names_make_unique()
# VDJ data
adata_vdj = ir.io.read_10x_vdj("/media/agschober/HDD12/3_scRNA-Seq_Sina/Cellranger_output/2nd_Experiment/run4/outs/per_sample_outs/run4/vdj_t/filtered_contig_annotations.csv")
#ir.pp.merge_with_ir(adata, adata_vdj)
# Epitope data
adata.uns['epitopes'] = epitope_ids
for e in epitope_ids:
adata.obs[e] = adata[:, e].X.A.copy()
# Hashtag data
adata.uns['hashtags'] = hashtags
for h in hashtags:
adata.obs[h] = adata[:, h].X.A.copy()
# CiteSeq Data
adata.uns['cite_ids'] = cite_seqs
for c in cite_seqs:
adata.obs[c] = adata[:, c].X.A.copy()
# Remove Barcodes from counts
adata = adata[:, [gene for gene in adata.var_names if gene not in feature_barcode_ids]]
adata.obs['sample'] = f'sample8'
adata.shape
</code>
<code>
#fuse the information of gene expression and tcr
adata = mu.MuData({"gex": adata, "airr": adata_vdj})
</code>
<code>
adata.shape
</code>
### Quality control
Basic analysis by amount counts, genes, and fraction of mitochondrial genes
<code>
adata["gex"].obs['n_counts'] = adata["gex"].X.A.sum(axis=1)
adata["gex"].obs['log_counts'] = np.log10(adata["gex"].obs['n_counts'])
adata["gex"].obs['n_genes'] = (adata["gex"].X.A > 0).sum(axis=1)
adata["gex"].obs['log_genes'] = np.log10(adata["gex"].obs['n_genes'])
mt_gene_mask = [gene.startswith('MT-') for gene in adata.var_names]
mt_gene_idx = np.where(mt_gene_mask)[0]
adata["gex"].obs['mt_frac'] = adata["gex"].X.A[:, mt_gene_idx].sum(1) / adata["gex"].X.A.sum(axis=1)
</code>
<code>
print('Mean # Genes: ', adata["gex"].obs['n_genes'].mean())
print('Median # Genes: ', adata["gex"].obs['n_genes'].median())
print('Mean # Counts: ', adata["gex"].obs['n_counts'].mean())
print('Median # Counts: ', adata["gex"].obs['n_counts'].median())
print('Mean % MT: ', adata["gex"].obs['mt_frac'].mean())
print('Median % MT: ', adata["gex"].obs['mt_frac'].median())
</code>
<code>
rcParams['figure.figsize'] = (4, 4)
sc.pl.violin(adata["gex"], ['n_counts'], size=1, log=False, rotation=90)
sc.pl.violin(adata["gex"], ['n_genes'], size=1, log=False, rotation=90)
sc.pl.violin(adata["gex"], ['mt_frac'], size=1, log=False, rotation=90)
</code>
- counts up to 25000, but mostly below 10000
- number of genes up to 6000, but mostly below 4000
- mitochondrial fraction up to 0.4 but mostly below 0.1
<code>
rcParams['figure.figsize'] = (8, 8)
sc.pl.scatter(adata["gex"], y='n_genes', x='n_counts', color ='mt_frac', size=10, show=False)
sc.pl.scatter(adata["gex"][np.logical_and(adata["gex"].obs['n_genes']<1500, adata["gex"].obs['n_counts']<8000)],
y='n_genes', x='n_counts', color='mt_frac', size=10, show=False)
plt.show()
</code>
<code>
b = ((adata['gex'].obs['n_counts']).sort_values()).to_list()
c = ((adata['gex'].obs['n_genes']).sort_values()).to_list()
</code>
<code>
plt.plot(b)
plt.ylabel('counts')
plt.xlabel('barcode')
</code>
<code>
plt.plot(c)
plt.ylabel('genes')
plt.xlabel('barcode')
</code>
<code>
plt.plot(b)
plt.ylabel('counts')
plt.xlabel('barcode')
plt.ylim((0,3000))
plt.xlim((0,200))
</code>
<code>
plt.plot(c)
plt.ylabel('genes')
plt.xlabel('barcode')
plt.ylim((0,1000))
plt.xlim((0,200))
</code>
- remove cells with more than 4000 genes and more than 12500 counts
- remove cells with more than 0.1 mt_fraction
- remove cells with less than 400 genes and 1200 counts
### Filtering of the cells
<code>
params_filter = { 'mt_frac': 0.1,
'n_counts_min': 1200,
'n_counts_max': 12500,
'n_genes_min': 400,
}
</code>
<code>
print(f'Size before filtering: {len(adata)}')
adata = adata[adata["gex"].obs['mt_frac'] < params_filter['mt_frac']]
adata = adata[adata["gex"].obs['n_counts'] > params_filter['n_counts_min']]
adata = adata[adata["gex"].obs['n_counts'] < params_filter['n_counts_max']]
adata = adata[adata["gex"].obs['n_genes'] > params_filter['n_genes_min']].copy()
print(f'Size after filtering: {len(adata)}')
adata.shape
</code>
### QC after filtering
<code>
rcParams['figure.figsize'] = (4, 4)
sc.pl.violin(adata["gex"], ['n_counts'], size=1, log=False, rotation=90)
sc.pl.violin(adata["gex"], ['n_genes'], size=1, log=False, rotation=90)
sc.pl.violin(adata["gex"], ['mt_frac'], size=1, log=False, rotation=90)
rcParams['figure.figsize'] = (8, 8)
sc.pl.scatter(adata["gex"], y='n_genes', x='n_counts', color ='mt_frac', size=10, show=False)
sc.pl.scatter(adata["gex"][np.logical_and(adata["gex"].obs['n_genes']<1500, adata["gex"].obs['n_counts']<8000)],
y='n_genes', x='n_counts', color='mt_frac', size=10, show=False)
plt.show()
</code>
### TCR stats
<code>
ir.pp.index_chains(adata)
ir.tl.chain_qc(adata)
</code>
<code>
adata.obs['airr:chain_pairing'].loc[(adata.obs['airr:chain_pairing']).isna()] = 'no_IR'
</code>
<code>
adata.obs['airr:chain_pairing'].value_counts()
</code>
<code>
def get_percentages_tcr(data):
df = ir.get.airr(data, "junction_aa", ["VJ_1", "VDJ_1", "VJ_2", "VDJ_2"])
p_alpha = df['VJ_1_junction_aa'].notnull().mean()
p_beta = df['VDJ_1_junction_aa'].notnull().mean()
p_paired = (df['VDJ_1_junction_aa'].notnull() & df['VJ_1_junction_aa'].notnull()).mean()
return [p_alpha, p_beta, p_paired]
chains = ['Alpha', 'Beta', 'Paired']
percentages = get_percentages_tcr(adata)
df_tcr_fractions = {
'chain': chains,
'percentage': percentages
}
df_tcr_fractions = pd.DataFrame(df_tcr_fractions)
g = sb.barplot(data=df_tcr_fractions, y='percentage', x='chain')
_ = g.set_xticklabels(rotation=30, labels=chains)
</code>
### Normalise
<code>
sc.pp.normalize_total(adata["gex"], target_sum=1e4)
sc.pp.log1p(adata["gex"])
</code>
### Quick Visual Sanity Check
<code>
utils_representation.calculate_umap(adata["gex"], n_high_var=5000, remove_tcr_genes=True)
</code>
<code>
adata["gex"].obs['chain_pairing'] = adata.obs['airr:chain_pairing']
</code>
<code>
sc.pl.umap(adata["gex"])
</code>
<code>
rcParams['figure.figsize'] = (6, 6)
sc.pl.umap(adata["gex"], color=['chain_pairing'])
</code>
<code>
sc.pl.umap(adata["gex"], color=['n_counts', 'log_counts', 'n_genes', 'mt_frac'], ncols=2)
</code>
### Separate the samples
<code>
utils_vis.distributions_over_columns(adata["gex"], hashtags, 2, 4)
</code>
<code>
def hash_solo_by_sample(hashtag_cols, col_name, n_noise_barcodes):
adata["gex"].obs[col_name] = 'NaN'
dfs_donor = []
adata["gex"].obs = adata["gex"].obs.drop(col_name, axis=1)
sc.external.pp.hashsolo(adata["gex"], hashtag_cols, number_of_noise_barcodes=n_noise_barcodes)
adata["gex"].obs = adata["gex"].obs.rename(columns={'Classification': col_name})
hash_solo_by_sample(hashtags, 'pool', 3)
adata["gex"].obs['pool'].value_counts()
</code>
<code>
hash_solo_by_sample(hashtags, 'pool', 5)
adata["gex"].obs['pool'].value_counts()
</code>
<code>
rcParams['figure.figsize'] = (16, 4)
for h in hashtags:
adata["gex"].obs[f'log_{h}'] = np.log(adata["gex"].obs[h].values+1)
sb.violinplot(data=adata["gex"].obs[[f'log_{h}' for h in hashtags]], scale='area')
</code>
<code>
utils_vis.adt_counts_by_condition(adata["gex"], hashtags, 'pool', 8, 4, do_log=True)
</code>
<code>
rcParams['figure.figsize'] = (8, 8)
sc.pl.umap(adata["gex"], color='pool')
</code>
<code>
rcParams['figure.figsize'] = (8, 8)
adata_ha = ann.AnnData(X=adata["gex"].obs[adata["gex"].uns['hashtags']], obs=adata["gex"].obs[['pool']])
adata_ha.var_names = adata["gex"].uns['hashtags']
sc.pp.log1p(adata_ha)
sc.pp.neighbors(adata_ha)
sc.tl.umap(adata_ha)
sc.pl.umap(adata_ha, color=['pool'] + [f'sample{i}' for i in range(1, 9)], ncols=3,
save=f'sample8_hashtag_umap.pdf')
</code>
<code>
adata = adata[~adata["gex"].obs['pool'].isin(['Doublet', 'Negative'])]
</code>
### Remove Epitope Counts
<code>
epitope_2_sample = {'NS4B214': ['sample1', 'sample2', 'sample3', 'sample4', 'sample5', 'sample6', 'sample7', 'sample8'],}
</code>
<code>
for e, samples in epitope_2_sample.items():
adata["gex"].obs.loc[~adata["gex"].obs['pool'].isin(samples), e] = np.nan
</code>
### Remove Totalseq Counts
<code>
samples_full_totalseq = ['sample1', 'sample2', 'sample3', 'sample4', 'sample5', 'sample6', 'sample7', 'sample8']
</code>
<code>
for c in cite_seqs:
adata["gex"].obs.loc[~adata["gex"].obs['pool'].isin(samples_full_totalseq), c] = np.nan
</code>
### Save
<code>
adata["gex"].obs['pool'] = f'sample8' + adata["gex"].obs['pool'].astype(str)
adata.write(filename="/media/agschober/HDD12/3_scRNA-Seq_Sina/Preprocessing/data8.h5mu")
</code>
<code>
import session_info
session_info.show()
</code>
|
{
"filename": "scRNAseq_Analysis_PartI_sample8.ipynb",
"repository": "SchoberLab/YF",
"query": "transformed_from_existing",
"size": 22277,
"sha": ""
}
|
# rag-qa_2.ipynb
Repository: dair-ai/maven-pe-for-llms-13
# Data-Augmented Question Answering
We are interested to build a personal learning assistant using LangChain. The parts we need:
- user question (input)
- role prompting to mimic learning assistant role
- relevant context obtained via data source
- knowledge base/data source (we are using lecture transcriptions for simplicity)
- vector database to store the data source and support semantic search
- personalized response with source/citations (summarized output)
<a href="https://colab.research.google.com/github/dair-ai/maven-pe-for-llms-9/blob/main/demos/session-4/rag-qa.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a>
<code>
%%capture
# update or install the necessary libraries
!pip install --upgrade openai
!pip install --upgrade langchain
!pip install --upgrade python-dotenv
!pip install --upgrade chromadb
</code>
<code>
import openai
import os
import IPython
from langchain.llms import OpenAI
from dotenv import load_dotenv
</code>
<code>
load_dotenv()
# API configuration
openai.api_key = os.getenv("OPENAI_API_KEY")
# for LangChain
os.environ["OPENAI_API_KEY"] = os.getenv("OPENAI_API_KEY")
</code>
First, we need to download the data we want to use as source to augment generation.
<code>
from langchain.embeddings.openai import OpenAIEmbeddings
from langchain.text_splitter import CharacterTextSplitter
from langchain.vectorstores.elastic_vector_search import ElasticVectorSearch
from langchain.vectorstores import Chroma
from langchain.docstore.document import Document
from langchain.prompts import PromptTemplate
from langchain.chains.qa_with_sources import load_qa_with_sources_chain
from langchain.llms import OpenAI
</code>
As our data source, we will use a transcription of Karpathy's recent lecture on GPT.
<code>
# split text into chunks
with open('../data/kar-gpt.txt') as f:
text_data = f.read()
text_splitter = CharacterTextSplitter(chunk_size=1000, chunk_overlap=0, separator=" ")
texts = text_splitter.split_text(text_data)
# embeddings obtained from OpenAI (you can use open-source like FAISS)
embeddings = OpenAIEmbeddings()
</code>
<code>
texts[:2]
</code>
<code>
docsearch = Chroma.from_texts(texts, embeddings, metadatas=[{"source": str(i)} for i in range(len(texts))])
</code>
<code>
query = "What is the course about?"
docs = docsearch.similarity_search(query)
</code>
<code>
chain = load_qa_with_sources_chain(OpenAI(temperature=0), chain_type="stuff")
query = "What is the course about?"
chain({"input_documents": docs, "question": query}, return_only_outputs=True)
</code>
<code>
template = """
Given the following extracted parts of a long document and a question, create a final answer with references ("SOURCES").
If you don't know the answer, just say that you don't know. Don't try to make up an answer.
ALWAYS return a "SOURCES" part in your answer.
=========
{summaries}
=========
Given the summary above, help answer the following question from the user:
Question: {question}
"""
# create a prompt template
PROMPT = PromptTemplate(template=template, input_variables=["summaries", "question"])
# query
chain = load_qa_with_sources_chain(OpenAI(temperature=0), chain_type="stuff", prompt=PROMPT)
query = "What is the course about?"
chain({"input_documents": docs, "question": query}, return_only_outputs=True)
</code>
Check out other chains like mapreduce and refine if you are working with bigger context and larger documents. https://docs.langchain.com/docs/components/chains/index_related_chains
<code>
from langchain import PromptTemplate, LLMChain
from langchain.chains import SimpleSequentialChain
llm = OpenAI(temperature=0.9)
response_prompt = PromptTemplate(
input_variables=["response"],
template="""You are a personal learning assistant.
Just take the answer from the previous response {response} and summarize it into one sentence.
Agent:
"""
)
query = "What is the course about?"
response_chain = ( {"response": chain} | response_prompt | llm)
response_chain.invoke({"input_documents": docs ,"question": query})
</code>
Exercise: Add another chain that connects with the previous `agent_chain` to create another agent that tries to be helpful and follows up with a question if it helps to keep the conversation going.
|
{
"filename": "rag-qa_2.ipynb",
"repository": "dair-ai/maven-pe-for-llms-13",
"query": "transformed_from_existing",
"size": 11935,
"sha": ""
}
|
# webscaping_1.ipynb
Repository: redashu/ML
<a href="https://colab.research.google.com/github/redashu/ML/blob/master/webscaping.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a>
<code>
from urllib import request # for downloading data from url
from bs4 import BeautifulSoup # for souping
# pointing to URL
url='https://en.wikipedia.org/wiki/Machine_learning'
url1='https://www.google.com/covid19/'
htmldata=request.urlopen(url)
#htmldata.read() # it will download data in html format
soupdata=BeautifulSoup(htmldata,'html5lib')
# html data , html parser --
# what is HTML parser -- is collection of html tags that can scrape data from particular tag like h1 , html , a , p
# now selecting a particular tag for data scrape
atagdata=soupdata.findAll('p')
# now converting data into string format from HTML format
import time
mydata=""
for i in atagdata:
mydata += i.text
print(mydata)
#print(atagdata)
</code>
|
{
"filename": "webscaping_1.ipynb",
"repository": "redashu/ML",
"query": "transformed_from_existing",
"size": 46000,
"sha": ""
}
|
# example_external_evaluation_pipelines.ipynb
Repository: langfuse/langfuse-docs
---
description: This notebook explains how to build an external evaluation pipeline to measure the performance of your production LLM application using Langfuse
category: Evaluation
---
# Evaluate Langfuse LLM Traces with an External Evaluation Pipeline
This cookbook explains how to build an external evaluation pipeline to measure the performance of your production LLM application using Langfuse.
As a rule of thumb, we encourage you to check first if the [evaluations in the Langfuse UI](https://langfuse.com/docs/scores/model-based-evals) cover your use case. If your needs go beyond these, you can still implement in Langfuse custom evaluation templates without code.
Consider implementing an external evaluation pipeline if you need:
- More control over **when** traces get evaluated. You could schedule the pipeline to run at specific times or responding to event-based triggers like Webhooks.
- Greater flexibility with your custom evaluations, when your needs go beyond what’s possible with the Langfuse UI
- Version control for your custom evaluations
- The ability to evaluate data using existing evaluation frameworks
If your use case meets any of this situations, let’s go ahead and implement your first external evaluation pipeline!
<iframe
width="100%"
className="aspect-[3230/2160] rounded mt-10"
src="https://www.youtube-nocookie.com/embed/rHfME8KDmIw?si=V4m8smxZ219AKmOU"
title="YouTube video player"
frameborder="0"
allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture; web-share"
referrerpolicy="strict-origin-when-cross-origin"
allowFullScreen
></iframe>
---
By the end of this cookbook, you’ll be able to:
- Create a synthetic dataset to test your models.
- Use the Langfuse client to gather and filter traces of previous model runs
- Evaluate these traces offline and incrementally
- Add scores to existing Langfuse traces
Conceptually, we will implement the following architecture:
```mermaid
sequenceDiagram
participant Application
participant Langfuse
participant Pipeline
actor User
Application ->> Langfuse: Ingest new traces
Langfuse ->> Pipeline: Fetch traces via SDK/API
Pipeline->>Pipeline: Run custom evaluation function/package
Pipeline ->> Langfuse: Add score to trace via SDK/API
Langfuse ->> User: Analyze evaluation scores via UI & API
```
---
**Note**: While we’re using a Jupyter notebook for this cookbook, in production you'd use your preferred orchestration tool. Just make sure to extract the code into a .py file and ensure all dependencies are available at runtime.
## (Prep-work) Loading synthetic traces to Langfuse
In this demo, we’ll build a mock application: a science communicator LLM that explains any topic in an engaging and approachable way.
Since we don’t have real user data, our first step is to create a synthetic dataset. We’ll generate a variety of potential questions that real users might ask. While this is a great way to kickstart your LLM development, collecting real user queries as soon as possible is invaluable.
You can get your Langfuse API keys [here](https://cloud.langfuse.com/) and OpenAI API key [here](https://platform.openai.com/api-keys)
_**Note:** This notebook utilizes the [Langfuse OTel Python SDK v3](https://langfuse.com/docs/sdk/python/sdk-v3). For users of [Python SDK v2](https://langfuse.com/docs/sdk/python/decorators), please refer to [our legacy notebook](https://github.com/langfuse/langfuse-docs/blob/366ec9395851da998d390eac4ab8c4dd2e985054/cookbook/example_external_evaluation_pipelines.ipynb)._
<code>
%pip install langfuse openai deepeval --upgrade
</code>
<code>
import os
# Get keys for your project from the project settings page: https://cloud.langfuse.com
os.environ["LANGFUSE_PUBLIC_KEY"] = "pk-lf-..."
os.environ["LANGFUSE_SECRET_KEY"] = "sk-lf-..."
os.environ["LANGFUSE_HOST"] = "https://cloud.langfuse.com" # 🇪🇺 EU region
# os.environ["LANGFUSE_HOST"] = "https://us.cloud.langfuse.com" # 🇺🇸 US region
# Your openai key
os.environ["OPENAI_API_KEY"] = "sk-proj-..."
</code>
Let's go ahead and generate a list of topic suggestions that we can later query to our application.
<code>
import openai
topic_suggestion = """ You're a world-class journalist, specialized
in figuring out which are the topics that excite people the most.
Your task is to give me 50 suggestions for pop-science topics that the general
public would love to read about. Make sure topics don't repeat.
The output must be a comma-separated list. Generate the list and NOTHING else.
The use of numbers is FORBIDDEN.
"""
output = openai.chat.completions.create(
messages=[
{
"role": "user",
"content": topic_suggestion
}
],
model="gpt-4o",
temperature=1
).choices[0].message.content
topics = [item.strip() for item in output.split(",")]
for topic in topics:
print(topic)
</code>
Great job! You now have a list of interesting topics users might ask about. Next, let's have our science communicator LLM handle those queries and add the results to Langfuse. To keep things simple, we’ll use Langfuse’s `@observe()` decorator. This decorator automatically monitors all LLM calls (generations) nested in the function. We’re also using the `langfuse` class to label and tag the traces, making it easier to fetch them later.
<code>
from langfuse import observe, get_client
langfuse = get_client()
prompt_template = """
You're an expert science communicator, able to explain complex topics in an
approachable manner. Your task is to respond to the questions of users in an
engaging, informative, and friendly way. Stay factual, and refrain from using
jargon. Your answer should be 4 sentences at max.
Remember, keep it ENGAGING and FUN!
Question: {question}
"""
@observe()
def explain_concept(topic):
langfuse.update_current_trace(
name=f"Explanation '{topic}'",
tags=["ext_eval_pipelines"]
)
prompt = prompt_template.format(question=topic)
return openai.chat.completions.create(
messages=[
{
"role": "user",
"content": prompt,
}
],
model="gpt-4o-mini",
temperature=0.6
).choices[0].message.content
for topic in topics:
print(f"Input: Please explain to me {topic.lower()}")
print(f"Answer: {explain_concept(topic)} \n")
</code>
Now you should see in the *Traces* section of the langfuse UI the traces you just added.
Remember, the goal of this tutorial is to show you how to build an external evaluation pipeline. These pipelines will run in your CI/CD environment, or be run in a different orchestrated container service. No matter the environment you choose, three key steps always apply:
1. **Fetch Your Traces**: Get your application traces to your evaluation environment
2. **Run Your Evaluations**: Apply any evaluation logic you prefer
3. **Save Your Results**: Attach your evaluations back to the Langfuse trace used for calculating them.
For the rest of the notebook, we'll have one goal:
---
🎯 Goal: ***Every day, at 5 am, our pipeline should evaluate 50 traces from the previous day***
---
## 1. Fetch Your Traces
Fetching traces from Langfuse is straightforward. Just set up the Langfuse client and use one of its functions to fetch the data. We'll take an incremental approach: first, we'll fetch the initial 10 traces and evaluate them. After that, we'll add our scores back into Langfuse and move on to the next batch of 10 traces. We'll keep this cycle going until we've processed a total of 50 traces.
The `fetch_traces()` function has arguments to filter the traces by tags, timestamps, and beyond. We can also choose the number of samples for pagination. You can find more about other methods to [query traces](https://langfuse.com/docs/query-traces) in our docs.
<code>
from langfuse import get_client
from datetime import datetime, timedelta
BATCH_SIZE = 10
TOTAL_TRACES = 50
langfuse = get_client()
now = datetime.now()
five_am_today = datetime(now.year, now.month, now.day, 5, 0)
five_am_yesterday = five_am_today - timedelta(days=1)
traces_batch = langfuse.api.trace.list(page=1,
limit=BATCH_SIZE,
tags="ext_eval_pipelines",
from_timestamp=five_am_yesterday,
to_timestamp=datetime.now()
).data
print(f"Traces in first batch: {len(traces_batch)}")
</code>
## 2. Run your evaluations
Langfuse can handle numerical, boolean and categorical (`string`) scores. Wrapping your custom evaluation logic in a function is often a good practice. Evaluation functions should take a `trace` as input and yield a valid score. Let's begin with a simple example using a categorical score.
### 2.1. Categoric Evaluations
When analyzing the outputs of your LLM applications, you may want to evaluate traits that are best defined qualitatively, such as sentiment, tonality or text complexity (Grade level).
We're building a science educator LLM that should sound engaging and positive.
To ensure it hits the right notes, we'll evaluate the tone of its outputs to see if they match our intent. We'll draft an evaluation prompt ourselves (no library) to identify the three main tones in each model output.
<code>
template_tone_eval = """
You're an expert in human emotional intelligence. You can identify with ease the
tone in human-written text. Your task is to identify the tones present in a
piece of <text/> with precission. Your output is a comma separated list of three
tones. PRINT THE LIST ALONE, NOTHING ELSE.
<possible_tones>
neutral, confident, joyful, optimistic, friendly, urgent, analytical, respectful
</possible_tones>
<example_1>
Input: Citizen science plays a crucial role in research by involving everyday
people in scientific projects. This collaboration allows researchers to collect
vast amounts of data that would be impossible to gather on their own. Citizen
scientists contribute valuable observations and insights that can lead to new
discoveries and advancements in various fields. By participating in citizen
science projects, individuals can actively contribute to scientific research
and make a meaningful impact on our understanding of the world around us.
Output: respectful,optimistic,confident
</example_1>
<example_2>
Input: Bionics is a field that combines biology and engineering to create
devices that can enhance human abilities. By merging humans and machines,
bionics aims to improve quality of life for individuals with disabilities
or enhance performance for others. These technologies often mimic natural
processes in the body to create seamless integration. Overall, bionics holds
great potential for revolutionizing healthcare and technology in the future.
Output: optimistic,confident,analytical
</example_2>
<example_3>
Input: Social media can have both positive and negative impacts on mental
health. On the positive side, it can help people connect, share experiences,
and find support. However, excessive use of social media can also lead to
feelings of inadequacy, loneliness, and anxiety. It's important to find a
balance and be mindful of how social media affects your mental well-being.
Remember, it's okay to take breaks and prioritize your mental health.
Output: friendly,neutral,respectful
</example_3>
<text>
{text}
</text>
"""
test_tone_score = openai.chat.completions.create(
messages=[
{
"role": "user",
"content": template_tone_eval.format(
text=traces_batch[1].output),
}
],
model="gpt-4o",
temperature=0
).choices[0].message.content
print(f"User query: {traces_batch[1].input['args'][0]}")
print(f"Model answer: {traces_batch[1].output}")
print(f"Dominant tones: {test_tone_score}")
</code>
Identifying human intents and tones can be tricky for language models. To handle this, we used a multi-shot prompt, which means giving the model several examples to learn from. Now let's wrap our code in an evaluation function for convenience.
<code>
def tone_score(trace):
return openai.chat.completions.create(
messages=[
{
"role": "user",
"content": template_tone_eval.format(text=trace.output),
}
],
model="gpt-4o",
temperature=0
).choices[0].message.content
tone_score(traces_batch[1])
</code>
Great! Now let's go ahead and create a numeric evaluation score.
### 2.2. Numeric Evaluations
In this cookbook, we'll use the `Deepeval` framework ([docs](https://docs.confident-ai.com/docs/getting-started)) to handle our numeric evaluations. Deepeval provides scores ranging from zero to one for many common LLM metrics. Plus, you can create custom metrics by simply describing them in plain language. To ensure our app's responses are joyful and engaging, we'll define a custom 'joyfulness' score.
You can use any evaluation library. These are popular ones:
- OpenAI Evals ([GitHub](https://github.com/openai/evals))
- Langchain Evaluators
- [RAGAS](https://docs.ragas.io/en/latest/concepts/metrics/index.html) for RAG applications
<code>
from deepeval.metrics import GEval
from deepeval.test_case import LLMTestCaseParams, LLMTestCase
def joyfulness_score(trace):
joyfulness_metric = GEval(
name="Correctness",
criteria="Determine whether the output is engaging and fun.",
evaluation_params=[LLMTestCaseParams.ACTUAL_OUTPUT],
)
test_case = LLMTestCase(
input=trace.input["args"],
actual_output=trace.output)
joyfulness_metric.measure(test_case)
print(f"Score: {joyfulness_metric.score}")
print(f"Reason: {joyfulness_metric.reason}")
return {"score": joyfulness_metric.score, "reason": joyfulness_metric.reason}
joyfulness_score(traces_batch[1])
</code>
Under the hood, GEval uses chain of thought (CoT) prompting to formulate a set of criteria for scoring prompts. When developing your own metrics, it's important to review the reasoning behind these scores. This helps ensure that the model evaluates the traces just as you intended when you wrote the evaluation prompt.
Our eval function returns a dictionary with both the score and the model's reasoning. We do this as we'll persist the reasoning with every langfuse score, ensuring interpretability.
Now we're done with defining our evaluation functions. Let's push those scores back to Langfuse!
## 3. Pushing Scores to Langfuse
Now that we have our evaluation functions ready, it’s time to put them to work. Use the Langfuse client to add scores to existing traces.
<code>
langfuse.create_score(
trace_id=traces_batch[1].id,
name="tone",
value=joyfulness_score(traces_batch[1])["score"],
comment=joyfulness_score(traces_batch[1])["reason"]
)
</code>
And thus, you've added your first externally-evaluated score to Langfuse! Just 49 more to go 😁. But don't worry — our solutions are easy to scale.
## 4. Putting everything together
Until now, we went through each of the necessary steps to build an external evaluation pipeline: Fetching traces, running the evaluations, and persisting the scores to Langfuse. Let's sum it up into a compact script that you could run in your evaluation pipeline.
We'll fetch the data in batches of 10 traces and then iterate through each trace to score it and push the scores back to Langfuse. Note that this batch size is for demonstration purposes. In a production setup, you might want to process multiple batches in parallel to speed things up. Batching not only reduces the memory load on your system but also allows you to create checkpoints, so you can easily resume if something goes wrong.
<code>
import math
for page_number in range(1, math.ceil(TOTAL_TRACES/BATCH_SIZE)):
traces_batch = langfuse.api.trace.list(
tags="ext_eval_pipelines",
page=page_number,
from_timestamp=five_am_yesterday,
to_timestamp=five_am_today,
limit=BATCH_SIZE
).data
for trace in traces_batch:
print(f"Processing {trace.name}")
if trace.output is None:
print(f"Warning: \n Trace {trace.name} had no generated output, \
it was skipped")
continue
langfuse.create_score(
trace_id=trace.id,
name="tone",
value=tone_score(trace)
)
jscore = joyfulness_score(trace)
langfuse.create_score(
trace_id=trace.id,
name="joyfulness",
value=jscore["score"],
comment=jscore["reason"]
)
print(f"Batch {page_number} processed 🚀 \n")
</code>
If your pipeline ran successfully, you should see your score in the Langfuse UI.
And that's it! You're now ready to integrate these lines into your preferred orchestration tool to ensure they run at the right times.
To achieve our original goal of running the script every day at 5 am, simply schedule a Cron task in your chosen environment with the rule `cron(0 5 * * ? *)`.
Thanks for coding along! I hope you enjoyed the tutorial and found it helpful.
|
{
"filename": "example_external_evaluation_pipelines.ipynb",
"repository": "langfuse/langfuse-docs",
"query": "transformed_from_existing",
"size": 198856,
"sha": ""
}
|
# manifest_1.ipynb
Repository: adityakakarla/finetuned-manifest-generation
<code>
from openai import OpenAI
openai_api_key = ''
client = OpenAI(api_key=openai_api_key)
</code>
<code>
def create_manifest(wrapper_script_fp, LSID, author, docker_image, repo, documentation_url, filepath, output_fp='output/manifest'):
with open(wrapper_script_fp, 'r') as file:
wrapper_script = file.read()
with open('data/example_manifest', 'r') as file:
example_manifest = file.read()
completion = client.chat.completions.create(
model="ft:gpt-4o-mini-2024-07-18:personal::B1TFUey0",
messages=[
{"role": "system", "content": """You are a genius GenePattern developer who writes manifest files based on wrapper scripts. You
will be fired for any mistakes you make"""},
{"role": "user", "content": f"""
Act as a senior software developer. I am going to describe a GenePattern manifest file, and then I would like you to create one based on information I provide. The first line is a "#" followed by the name of the function I provide. The second line is a # followed by the current date and time. Do not include any blank lines in the output.
The next section should be pasted verbatim:
---------------------------------------------------------
JVMLevel=
LSID=
author=GenePattern Team + ChatGPT
commandLine=
cpuType=any
The next line should be "description=" followed by a brief description of the function I provided.
The next line should be "documentationUrl=" followed by the URL to the site where the function is described.
The next line should be "fileFormat=" followed by a comma-separated list of the file extensions output by the function.
The next line should be "job.cpuCount="
The next line should be "job.docker.image=[DOCKER IMAGE HERE]"
The next section should be pasted verbatim:
job.memory=
job.walltime=
language=any
The next line should start with "categories=" and should be most applicable term in following list: alternative splicing,batch correction,clustering,cnv analysis,data format conversion,differential expression,dimension reduction,ecdna,flow cytometry,gene list selection,gsea,image creators,metabolomics,methylation,missing value imputation,mutational significance analysis,pathway analysis,pipeline,prediction,preprocess & utilities,projection,proteomics,rna velocity,rna-seq,rnai,sage,sequence analysis,single-cell,snp analysis,statistical methods,survival analysis,variant annotation,viewer,visualizer
The next line should be "name=" and then the name of the function.
The next line should be "os=any"
After that, identify all the parameters in the wrapper script.
---------------------------------------------------------
The manifest file should include each parameter of the provided function, in the format. I will provide below. Here are some instructions for this format:
1. When you see a # character, replace it with the number of the parameter in the provided function.
2. When you see "default_value=", place the parameter's default value after the "=" if there is one.
3. When you see "description=", add the parameter's description after the "="
4. When you see "name=", add the parameter's name after the "="
5. When you see "optional=", write "on" if the parameter is optional
6. In the parameter name, replace the "#" with the cardinal number of the parameter
7. When you see "flag=", add the parameter's command-line flag after the "=" if there is one. If there are more than one way of specifying a flag, use the one that starts with two hyphens: "--"
8. When you see "type=", add the parameter's type after the "=". The type should be the term in the following comma-separated list that corresponds most closely to the type of parameter: CHOICE,FILE,Floating Point,Integer,TEXT,java.lang.String. Pick java.io.File if you think the input are filepaths.
9. When you see "taskType=", add the type of analysis this module performs. For example: batch correction, visualizer, scRNA analysis. You can infer the category based on the name and description.
10 If you think a parameter is a file path, put IN for the p#_MODE
11. Order from p1-pn
12. Enter two new line characters between parameter info (ex: new line between p1_... and p2_...)
13. Ensure the manifest valid java.properties file and follows this syntax
14. Compare different types of parameters against known equivalents - java.lang.String vs text, etc.
---------------------------------------------------------
Here is the format for each parameter:
p#_MODE=
p#_TYPE=
p#_default_value=
p#_description=
p#_fileFormat=
p#_flag=
p#_name=
p#_numValues=
p#_optional=
p#_prefix=
p#_prefix_when_specified=
p#_type=
p#_value=
taskType=
Here is an example: {example_manifest}
---------------------------------------------------------
For the commandline, generate a Rscript commandline to run the wrapper script. The parameters should be: --flag <value>.
Relevant info:
LSID: {LSID}
author: {author}
docker_image: {docker_image}
repo: {repo}
documentation_url: {documentation_url},
filepath: {filepath}
wrapper_script: {wrapper_script}"""}
])
manifest = completion.choices[0].message.content
with open(output_fp, 'w') as file:
file.write(manifest)
file.close()
return manifest
</code>
<code>
output = create_manifest(wrapper_script_fp='input/wrapper.R',
LSID='urn:lsid:genepattern.org:module.analysis:00465:999999999',
author='Thorin Tabor;UCSD - Mesirov Lab',
docker_image='genepattern/spatialge-stgradient:0.2',
repo='https://github.com/genepattern/spatialGE.STgradient',
documentation_url='https://genepattern.github.io/spatialGE.STgradient/v1/',
filepath='/spatialGE/wrapper.R',)
</code>
|
{
"filename": "manifest_1.ipynb",
"repository": "adityakakarla/finetuned-manifest-generation",
"query": "transformed_from_existing",
"size": 7937,
"sha": ""
}
|
# Lecture 09_1.ipynb
Repository: ambujtewari/stats607a-fall2014
|
{
"filename": "Lecture 09_1.ipynb",
"repository": "ambujtewari/stats607a-fall2014",
"query": "transformed_from_existing",
"size": 100963,
"sha": ""
}
|
# second_level_evaluation-checkpoint_1.ipynb
Repository: thomyks/Automatic-Topic-Extraction-with-BERTopic
<code>
# Topic Diversity
# Describe the details.
</code>
<code>
import pandas as pd
from nltk.corpus import stopwords
from nltk.tokenize import word_tokenize
import nltk
# Ensure NLTK resources are downloaded
nltk.download('punkt')
nltk.download('stopwords')
# Load your dataset
file_path = 'Merged_BERTopic_Result_Reverse.csv' # Replace with your file path
data = pd.read_csv(file_path)
</code>
<code>
data_filtered = data[data['Topic Label'] != -1]
# Example: Assuming you have a separate list of documents
documents = data['text']
# Preprocess documents
stop_words = set(stopwords.words('english'))
def preprocess(text):
tokens = word_tokenize(text.lower())
tokens = [token for token in tokens if token.isalpha() and token not in stop_words]
return tokens
processed_docs = [preprocess(doc) for doc in documents]
# Extract topics after filtering
topic_representations = data.groupby('Higher_Topic_Label')['Representation'].apply(lambda x: x.iloc[0].split(', '))
topics = topic_representations.tolist()
# Calculate Topic Diversity
def calculate_topic_diversity(topics):
unique_words = set()
total_words = 0
for topic in topics:
unique_words.update(topic) # Add words to the unique set
total_words += len(topic) # Count total words in all topics
# Topic diversity is the proportion of unique words to total words
topic_diversity = len(unique_words) / total_words
return topic_diversity
# Calculate topic diversity
topic_diversity = calculate_topic_diversity(topics)
# Display the topic diversity score
print(f"\nTopic Diversity: {topic_diversity}")
</code>
Topic Coherence
<code>
# Function to remove brackets and quotes, keeping only the words
def clean_column(text):
# Remove square brackets and split by commas
return text.strip("[]").replace("'", "")
# Apply the function to the DataFrame column
data['Representation'] = data['Representation'].apply(clean_column)
data
</code>
<code>
from gensim.models import CoherenceModel
from gensim.corpora import Dictionary
import pandas as pd
# Assuming 'processed_docs' is a list of tokenized documents
# Create a Gensim dictionary from the processed documents
dictionary = Dictionary(processed_docs)
per_topic_coherence_cv = []
for topic in topics:
# Create a list containing just the current topic
current_topic = [topic]
print(current_topic)
# print(processed_docs)
# print(dictionary)
# Initialize the CoherenceModel for the current topic using 'c_v'
coherence_model_cv = CoherenceModel(topics=current_topic, texts=processed_docs, dictionary=dictionary, coherence='c_v')
# Compute the c_v coherence score
coherence_cv = coherence_model_cv.get_coherence()
print(coherence_cv)
# Append the c_v score to the list
per_topic_coherence_cv.append(coherence_cv)
# Create a DataFrame to display the results
results = pd.DataFrame({
"Keywords": topic_representations.values,
"Coherence c_v": per_topic_coherence_cv,
})
# Display overall and per-topic coherence scores
print(results)
results.to_csv('Second_Level_Topic_coherence.csv')
</code>
<code>
# Create a DataFrame to display the results
results = pd.DataFrame({
"Keywords": topic_representations.values,
"Coherence c_v": per_topic_coherence_cv,
})
# Display overall and per-topic coherence scores
print(results)
results.to_csv('Second_Level_Coherence.csv')
</code>
<code>
# Create a DataFrame from the list
df = pd.DataFrame(per_topic_coherence_cv, columns=['Topic Coherence Score'])
# Convert to CSV
df.to_csv('per_topic_coherence_cv.csv', index='Index')
</code>
<code>
file_path = 'Second_Level_Coherence.csv' # Replace with your file path
data = pd.read_csv(file_path)
mean_score = data['Coherence c_v'].mean()
data['Coherence c_v'].mean()
mean_score
</code>
|
{
"filename": "second_level_evaluation-checkpoint_1.ipynb",
"repository": "thomyks/Automatic-Topic-Extraction-with-BERTopic",
"query": "transformed_from_existing",
"size": 88655,
"sha": ""
}
|
# gcn_1.ipynb
Repository: dcolinmorgan/gcn
<code>
%reset -f
%config Completer.use_jedi = True
# %matplotlib widget
# from scipy.stats import rankdata
# from sklearn.preprocessing import normalize
# import sklearn.utils as sku
# import plotly.graph_objects as go
# import plotly.express as px
# import chart_studio.plotly as py
# import chart_studio
# chart_studio.tools.set_credentials_file(username='dcolinmorgan', api_key='9FS3nO6nWYFq5zT6BRHD')
import plotly
import scipy
import warnings
warnings.filterwarnings('ignore')
##network stuff
from algorithmx import jupyter_canvas
import graph_tool.all as gt
from graph_tool import *
from graph_tool.all import *
from pylab import *
import algorithmx
import networkx as nx
import leidenalg as la
import igraph as ig
import community as community_louvain
import networkx.algorithms.community as nx_comm
from networkx.algorithms import bipartite
import graphistry
graphistry.register(api=3, username='dcolinmorgan', password='f5UwthGEF@F@xnP')
</code>
<code>
import matplotlib.pyplot as plt
import numpy as np
import os,glob,sys,importlib,pickle,tqdm
from itertools import combinations,chain#,scipy,coolbox,pybedtools,
# from scipy.stats import linregress
# from scipy.ndimage import gaussian_filter
from tqdm import tqdm
from IPython.display import Image
import pandas as pd
import seaborn as sns
from scipy import stats
from pathlib import Path
# import pyvis
# from pyvis.network import Network
import networkit as nk
# from statannot import add_stat_annotation
from statannotations.Annotator import Annotator
# import biosppy
# from sklearn import metrics
os.chdir('/home/dcmorgan')
os.getcwd()
</code>
<code>
patt='all'
sys.path.insert(1, './run/gcn/')
import gcn_func
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts, meas, time_bar,proc_dat,rev_tbar,group_time_plot,time_order_net,build_gcn,structural_analysis
from gcn_func import pdize_net, plot_comm, load_list_of_dicts,igraph_from_pandas
# import mygene
# mg = mygene.MyGeneInfo()
from biothings_client import get_client
gene_client = get_client('gene')
sys.path.insert(1, './nestedness_analysis/')
import nestedness_metrics_other_functions
from nestedness_metrics_other_functions import from_edges_to_matrix
# importlib.reload(sys.modules['EO_functions_bipartite'])
import extremal_bi
sys.path.insert(1, './nestedness_analysis/network-generator/')
import generate_synthetic_networks
from netgen import NetworkGenerator
</code>
# bioinfo > net
<code>
R0396MWT=pd.read_csv('hmp_subset_genefamilies-cpm.tsv',sep='\t',usecols=[0,1])
R0396MWT=R0396MWT[R0396MWT['R0003-CPK-DNA_Abundance-RPKs']!=0]
</code>
<code>
# R0396MWT['gene']=R0396MWT['# Gene Family'].str.split('|').str[0].str.split('_').str[1].tolist()
# R0396MWT['spec']=R0396MWT['# Gene Family'].str.split('|').str[1].tolist()
# R0396MWT=R0396MWT[['spec','gene']]
# R0396MWT.spec=R0396MWT.spec.astype('str')
R0396MWTa=R0396MWT.loc[np.random.choice(R0396MWT.index, 5000, replace=False)]
</code>
<code>
[len(np.unique(R0396MWTa['spec'])),
len(np.unique(R0396MWTa['gene'].astype('str')))]
</code>
<code>
G=nx.from_pandas_edgelist(R0396MWTa, 'gene', 'spec', edge_attr=None, create_using=None)
# G = nx.Graph(source=['gene'],target=R0396MWT['spec'])
</code>
<code>
from networkx.algorithms import bipartite
# G = nx.path_graph(4)
print(bipartite.is_bipartite(G))
# is_bipartite(G)
</code>
<code>
g = nk.generators.HyperbolicGenerator(1e5).generate()
communities = nk.community.detectCommunities(g, inspect=True)
</code>
<code>
%%bash
pat=R0396-MWT
awk '($3>90)' run/gcn/pat/"$pat"_proteins_o2 > "$pat"_nko
cut -f2 "$pat"_nko |cut -d "|" -f 1|cut -d "_" -f 2 > "$pat"_tmpA
cut -f2 "$pat"_nko |cut -d "|" -f 2 > "$pat"_tmpB
cut -f3 "$pat"_nko >"$pat"_tmpC
pr -mt -s, "$pat"_tmpB "$pat"_tmpA "$pat"_tmpC >"$pat"_nk
# awk '($3>90)' "$pat"_tmpD >"$pat"_nk
rm *tmp*
</code>
<code>
reader = nk.graphio.EdgeListReader(',',1,'#',directed=False,continuous=False)
G = reader.read('R0396-MWT_nk')
map = reader.getNodeMap()
</code>
<code>
dd = sorted(nk.centrality.DegreeCentrality(G).run().scores(), reverse=True)
plt.xscale("log")
plt.xlabel("degree")
plt.yscale("log")
plt.ylabel("number of nodes")
plt.plot(dd)
plt.show()
</code>
<code>
nk.profiling.Profile.create(G).output('pdf','tmp')
</code>
<code>
plmCommunities = nk.community.detectCommunities(G, algo=nk.community.PLM(G, True))
</code>
<code>
print("{0} elements assigned to {1} subsets".format(plmCommunities.numberOfElements(),
plmCommunities.numberOfSubsets()))
nk.community.writeCommunities(plmCommunities, "tmp/communtiesPLM.partition")
</code>
<code>
sizes = communities.subsetSizes()
sizes.sort(reverse=True)
ax1 = plt.subplot(2,1,1)
ax1.set_ylabel("size")
ax1.plot(sizes)
ax2 = plt.subplot(2,1,2)
ax2.set_xscale("log")
ax2.set_yscale("log")
ax2.set_ylabel("size")
ax2.plot(sizes)
plt.show()
</code>
<code>
coreDec = nk.centrality.CoreDecomposition(g)
coreDec.run()
nk.viztasks.drawGraph(g, node_size=[(k**2)*20 for k in coreDec.scores()])
plt.show()
</code>
# pheno
<code>
primary=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='Primary Data')
diet=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='Diet Data')
blood_stool=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='blood and stool biomarkers')
secondary=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='Secondary Data')
MRI=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='MRI scores')
</code>
<code>
uni_bact=primary[['Age','Hypertension Category by 24h BP w/o considering antihypertensive med']]
uni_bact=uni_bact.rename(columns={"Hypertension Category by 24h BP w/o considering antihypertensive med": "HT"})
# uni_bact.to_csv('data/gcn/uni_bact.txt',sep='\t')
</code>
# build networks
<code>
# %%bash ARG
# humann_join_tables -i /groups/cgsd/gordonq/LeungWK_Metagenomics_SS-190611-01a/humann3 -o arg_subset_genefamilies.tsv --file_name genefamilies
# humann_renorm_table -i arg_subset_genefamilies.tsv -o arg_subset_genefamilies-cpm.tsv --units cpm
</code>
<code>
# build_gcn('arg_subset_genefamilies-cpm.tsv','ARG')
</code>
<code>
# %%bash ##CRC
# humann_join_tables -i /groups/cgsd/gordonq/CPOS_Data*/LeungSY_Metagenomics_CPOS-*/humann3 -o crc_subset_genefamilies.tsv --file_name genefamilies
# humann_renorm_table -i crc_subset_genefamilies.tsv -o crc_subset_genefamilies-cpm.tsv --units cpm
</code>
<code>
# relab=pd.read_csv('../../groups/cgsd/gordonq/all_hypertension/342_K2_contigsum.csv')
# relab[['p', 'c','o','f','g','s']]=pd.DataFrame(relab["Unnamed: 0"].str.split(';', expand=True).values,
# columns=['p', 'c','o','f','g','s'])
# del relab["Unnamed: 0"]
# relgene=pd.read_csv('/groups/cgsd/gordonq/LauG_Metagenomics_CPOS-200710-CJX-3455a/50_genefamilies.tsv',sep='\t')
# relgene=pd.read_csv('50_genefamilies-cpm.tsv')
relgene=pd.read_csv('all_arg_subset_genefamilies-cpm.tsv',sep='\t')
# relgene=pd.read_csv('hmp_subset_genefamilies-cpm.tsv',sep='\t',nrows=100)
# relgene=pd.read_csv('',sep='\t')
relgene['gene']=relgene['# Gene Family'].str.split('|').str[0]
relgene=relgene[relgene['gene']!='UniRef90_unknown']
relgene=relgene[relgene['gene']!='UNMAPPED']
relgene.index=relgene['# Gene Family']
del relgene['gene'], relgene['# Gene Family']
# relgene=relgene/relgene.sum(axis=0)
# relgene=relgene/relgene.sum(axis=0)
relgene['gen']=relgene.index.str.split('|').str[1].str.split('.').str[0].tolist()
relgene['spec']=relgene.index.str.split('.').str[1]#.str.split('.').str[0].tolist()
relgene['spec'].replace('_',' ')
relgene.index=relgene.index.str.split('|').str[0]
relgene=relgene.dropna()
# del relgene['# Gene Family']
</code>
<code>
cc=relgene.groupby(['# Gene Family','spec']).sum()
# dd=relgene.groupby(['# Gene Family','gen']).sum()
cc=cc.reset_index()
# dd=dd.reset_index()
cc=cc.rename(columns={'# Gene Family':'gene'})#,'spec':0,'gene':1})
dd=cc[['gene','spec',net]]
dd=dd[dd[net]!=0]
</code>
check instrucitons [from here](https://stackoverflow.com/questions/23975773/how-to-compare-directed-graphs-in-networkx#54677502)
and [here](https://stackoverflow.com/questions/18261587/python-networkx-remove-nodes-and-edges-with-some-condition)
<code>
dd=cc[['spec','gene',net]]
dd=dd[dd[net]!=0]
B = nx.Graph()
dd=dd[dd['source'].str.contains('s__')]
dd=dd[dd['target'].str.contains('UniRef')]
B.add_nodes_from(dd['spec'], bipartite=0)
B.add_nodes_from(dd['gene'], bipartite=1)
B.add_edges_from(tuple(dd[['spec','gene']].itertuples(index=False, name=None)))
remove = [node for node,degree in dict(B.degree()).items() if degree <5]
B.remove_nodes_from(remove)
C.append(B)
</code>
<code>
from networkx.algorithms import bipartite
ff=[]
C=[]
for i,net in enumerate(relgene.columns[1:4]):
# pd.read_csv()
# dd=cc[['spec','gene',net]]
# dd=dd[dd[net]!=0]
# ee=nx.from_pandas_edgelist(dd,source='spec',target='gene',edge_attr=net)
# remove = [node for node,degree in dict(ee.degree()).items() if degree <5]
# ee.remove_nodes_from(remove)
# ff.append(ee)
B = nx.Graph()
dd=dd[dd['source'].str.contains('s__')]
dd=dd[dd['target'].str.contains('UniRef')]
B.add_nodes_from(dd['spec'], bipartite=0)
B.add_nodes_from(dd['gene'], bipartite=1)
B.add_edges_from(tuple(dd[['spec','gene']].itertuples(index=False, name=None)))
remove = [node for node,degree in dict(B.degree()).items() if degree <5]
B.remove_nodes_from(remove)
C.append(B)
# with open('data/gcn/NX_Emore_ARG.pkl', 'wb') as f:
# pickle.dump(ff, f)
with open('data/gcn/BXX_Emore_ARG.pkl', 'wb') as f:
pickle.dump(C, f)
</code>
<code>
graphs = load_list_of_dicts('data/gcn/BX_Emore_ARG.pkl')
len(graphs[12].edges())
# B=graphs[3]
# remove = [node for node,degree in dict(B.degree()).items() if degree <2]
# B.remove_nodes_from(remove)
# len(B.edges)
</code>
<code>
measur=eval('nx.degree_centrality')
tmp=plt.hist(measur(graphs[1]),bins=20)
tmp=plt.hist(measur(graphs[0]),bins=20,alpha=.5)
</code>
<code>
HT50=uni_bact[uni_bact.index.isin(relgene.columns[:-2].str.split('-').str[0])]
HT50['index']=np.arange(len(HT50))
S = [nx.clustering(graphs[i]) for i in HT50[HT50['HT']==0]['index'].values]
T = [nx.clustering(graphs[i]) for i in HT50[HT50['HT']!=0]['index'].values]
</code>
<code>
non=pd.DataFrame(S).melt()
non['type']='NoHT'
non.dropna(inplace=True)
non=non[non.value!=0]
non=non[~non['variable'].str.contains('UniRef90')]
non.value=non.value/np.sum(non.value)
yes=pd.DataFrame(T).melt()
yes['type']='HT'
yes.dropna(inplace=True)
yes=yes[yes.value!=0]
yes=yes[~yes['variable'].str.contains('UniRef90')]
yes.value=yes.value/np.sum(yes.value)
df=non.append(yes)
# df=df.dropna()
df['gen']=df.variable.str.split('_').str[2]
</code>
<code>
[sum(non.value),sum(yes.value)]
</code>
<code>
tmp=plt.hist(non.value,log=True,bins=100)
tmp=plt.hist(yes.value,log=True,bins=100,alpha=.5)
</code>
<code>
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
sns.set_theme(style="white", rc={"axes.facecolor": (0, 0, 0, 0)})
# Create the data
# rs = np.random.RandomState(1979)
# x = rs.randn(500)
# g = np.tile(list("ABCDEFGHIJ"), 50)
# df = pd.DataFrame(dict(x=x, g=g))
# m = df.g.map(ord)
# df["x"] += m
# Initialize the FacetGrid object
pal = sns.cubehelix_palette(10, rot=-.25, light=.7)
g = sns.FacetGrid(df[df['type']=='HT'], row="gen", hue="gen", aspect=20, height=.5, palette=pal)
# Draw the densities in a few steps
g.map(sns.kdeplot, "value",
bw_adjust=.5, clip_on=False,
fill=True, alpha=1, linewidth=1.5)
g.map(sns.kdeplot, "value", clip_on=False, color="w", lw=2, bw_adjust=.5)
# passing color=None to refline() uses the hue mapping
g.refline(y=0, linewidth=2, linestyle="-", color=None, clip_on=False)
# Define and use a simple function to label the plot in axes coordinates
def label(x, color, label):
ax = plt.gca()
ax.text(0, .2, label, fontweight="bold", color=color,
ha="left", va="center", transform=ax.transAxes)
g.map(label, "value")
# Set the subplots to overlap
g.figure.subplots_adjust(hspace=-.25)
# Remove axes details that don't play well with overlap
g.set_titles("")
g.set(yticks=[], ylabel="")
g.despine(bottom=True, left=True)
plt.savefig("data/gcn/degree_centrality_HT.png",dpi=300,bbox_inches = "tight")
</code>
<code>
relgene=pd.read_csv('hmp_subset_genefamilies-cpm.tsv',sep='\t')
relgene['gene']=relgene['# Gene Family'].str.split('|').str[0]
relgene=relgene[relgene['gene']!='UniRef90_unknown']
relgene=relgene[relgene['gene']!='UNMAPPED']
relgene.index=relgene['# Gene Family']
del relgene['gene'], relgene['# Gene Family']
# relgene=relgene/relgene.sum(axis=0)
relgene=relgene/relgene.sum(axis=0)
relgene['gen']=relgene.index.str.split('|').str[1].str.split('.').str[0].tolist()
relgene['spec']=relgene.index.str.split('.').str[1]#.str.split('.').str[0].tolist()
relgene['spec'].replace('_',' ')
relgene.index=relgene.index.str.split('|').str[0]
relgene=relgene.dropna()
cc=relgene.groupby(['# Gene Family','spec']).sum()
# dd=relgene.groupby(['# Gene Family','gen']).sum()
cc=cc.reset_index()
# dd=dd.reset_index()
cc=cc.rename(columns={'# Gene Family':'gene'})#,'spec':0,'gene':1})
primary=pd.read_excel('data/Data Raw - Gut Microbiome Cohort Project Database - 300 Cohort v3.0_280921.xlsx',index_col=0,sheet_name='Primary Data')
uni_bact=primary[['Age','Hypertension Category by 24h BP w/o considering antihypertensive med']]
uni_bact=uni_bact.rename(columns={"Hypertension Category by 24h BP w/o considering antihypertensive med": "HT"})
</code>
# analyze HT nets
<code>
patt='all'
sys.path.insert(1, './run/gcn/')
import gcn_func
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts, meas, time_bar,proc_dat,rev_tbar,group_time_plot,time_order_net,build_gcn#plotRidge,LayeredNetworkGraph,plot_sankey
# relgene=pd.read_csv('data/gcn/relgene_all.txt',sep='\t')
graphs = load_list_of_dicts('data/gcn/BX_'+patt+'_HT.pkl')
HTXX=uni_bact[uni_bact.index.isin(relgene.columns[1:-2].str.split('-').str[0])]
HTXX['index']=np.arange(len(HTXX))
</code>
<code>
# for i,net in tqdm.tqdm(enumerate(BX_graphs)):
for i,net in tqdm(enumerate(HTXX[HTXX['HT']!=5]['index'].values)):
cc=nx.convert_matrix.to_pandas_edgelist(graphs[i])
# cc['weight']=np.random.randn(len(cc))
rrr=str(HTXX[HTXX['index']==i]['Age'].item())+'_'+str(HTXX[HTXX['index']==i]['HT'].item())#+'_'+str(HTXX[HTXX['index']==i]['sex'].item())
cc.rename(columns={cc.columns[2]:rrr},inplace=True)
if i==0:
dd=cc
else:
dd=dd.merge(cc,on=['source','target'],how='outer')
# dd.dropna(how='any')
</code>
<code>
dd.set_index(['source', 'target'], inplace=True) #>> run only first time editing dd
# dd = dd/np.max(dd,axis=0)
# dd=dd/np.sum(dd,axis=0) ###tried to do things strangely
### dd=np.argsort(dd)
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
# [noHT.shape,HT1.shape,HT2.shape]
[noHT.shape,HT.shape]
</code>
## case v control nets
<code>
ccc=pd.DataFrame(noHT.sum(axis=1)).reset_index()
g=nx.from_pandas_edgelist(ccc,source='source',target='target',edge_attr=0)
C = nx.Graph()
C.add_nodes_from(ccc['source'], bipartite=0)
C.add_nodes_from(ccc['target'], bipartite=1)
C.add_weighted_edges_from(tuple(ccc[['source','target',0]].itertuples(index=False, name=None)))
ddd=pd.DataFrame(HT.sum(axis=1)).reset_index()
g=nx.from_pandas_edgelist(ddd,source='source',target='target',edge_attr=0)
B = nx.Graph()
B.add_nodes_from(ccc['source'], bipartite=0)
B.add_nodes_from(ccc['target'], bipartite=1)
B.add_weighted_edges_from(tuple(ccc[['source','target',0]].itertuples(index=False, name=None)))
pd.DataFrame(nx.degree_histogram(B))
pd.DataFrame(nx.degree_histogram(C))
tmp=plt.hist(nx.degree_histogram(C),log=True,bins=10)
tmp=plt.hist(nx.degree_histogram(B),log=True,bins=10)
</code>
<code>
ccc=pd.DataFrame(noHT.sum(axis=1)).reset_index()
# ccc=ccc[ccc[0]>10**-2].dropna()#percentage cutoff
ccc=ccc[ccc[0]>10].dropna()
# g=nx.from_pandas_edgelist(ccc,source='source',target='target',edge_attr=0)
C = nx.Graph()
C.add_nodes_from(ccc['source'], bipartite=0)
C.add_nodes_from(ccc['target'], bipartite=1)
C.add_weighted_edges_from(tuple(ccc[['source','target',0]].itertuples(index=False, name=None)))
ddd=pd.DataFrame(HT.sum(axis=1)).reset_index()
# ddd=ddd[ddd[0]>10**-2].dropna()
ddd=ddd[ddd[0]>10].dropna()
# g=nx.from_pandas_edgelist(ddd,source='source',target='target',edge_attr=0)
B = nx.Graph()
B.add_nodes_from(ddd['source'], bipartite=0)
B.add_nodes_from(ddd['target'], bipartite=1)
B.add_weighted_edges_from(tuple(ddd[['source','target',0]].itertuples(index=False, name=None)))
</code>
<code>
g = graphistry.edges(ddd, 'source', 'target','value')
g.plot()
</code>
<code>
g = graphistry.edges(ccc, 'source', 'target','value')
g.plot()
</code>
## bin & time steps
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
[noHT.shape,HT.shape]
noHT.columns=noHT.columns.str.split('_').str[0]
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT=noHT.dropna(how='any')
noHT.reset_index(inplace=True)
jj=noHT.melt(['source','target'])
jj.rename(columns={'variable':'time'},inplace=True)
jj['t']=jj['time']
jj['species']=jj['source'].str.split('_').str[2]
jj=jj.dropna(how='any')
sns.lineplot(data=jj[jj['target']=='UniRef90_T4BVE4'], x="t", y="value", hue="species")
</code>
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
[noHT.shape,HT.shape]
noHT.columns=noHT.columns.str.split('_').str[0]
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT=noHT.dropna(how='any')
noHT.columns=pd.qcut((noHT.columns).astype('int'), 10, labels=False)
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT.reset_index(inplace=True)
jj=noHT.melt(['source','target'])
jj.rename(columns={'variable':'time'},inplace=True)
jj['t']=jj['time']
jj['species']=jj['source'].str.split('_').str[2]
jj=jj.dropna(how='any')
sns.lineplot(data=jj[jj['target']=='UniRef90_T4BVE4'], x="t", y="value", hue="source")
</code>
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
[noHT.shape,HT.shape]
noHT.columns=noHT.columns.str.split('_').str[0]
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT=noHT.dropna(how='any')
noHT.columns=pd.qcut((noHT.columns).astype('int'), 5, labels=False)
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT.reset_index(inplace=True)
jj=noHT.melt(['source','target'])
jj.rename(columns={'variable':'time'},inplace=True)
jj['t']=jj['time']
jj['species']=jj['source'].str.split('_').str[2]
jj=jj.dropna(how='any')
sns.lineplot(data=jj[jj['target']=='UniRef90_T4BVE4'], x="t", y="value", hue="source")
</code>
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
TU,TD,noHTT,HTT=time_order_net(noHT,HT,10**-6,'target',5,1,'mean')
TU.reset_index(inplace=True)
TD.reset_index(inplace=True)
[TU.shape,TD.shape]
</code>
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
LU,LD,noHDL,HTL=time_order_net(noHT,HT,10**-6,['source','target'],5,1,'median')
LU.reset_index(inplace=True)
LD.reset_index(inplace=True)
[LU.shape,LD.shape]
</code>
<code>
# try to see which species are causing flux in protein content
# np.unique(noHTT[noHTT['target'].isin(LU[~LU['target'].isin(TU.index)]['target'])]['source'])
</code>
<code>
LinkUP=LU[LU['target'].isin(LD['target'])]#['source']
</code>
<code>
LinkDN=LD[LD['target'].isin(LU['target'])]#['source']
</code>
<code>
JEFF=LinkUP.append(LinkDN)#
JEFF[5]=JEFF[4]-JEFF[0]
</code>
<code>
JEFF
</code>
<code>
ax = sns.barplot(x="target", y=5, hue="source", data=JEFF[JEFF['target']=='UniRef90_T4BVE4'])
</code>
<code>
TU[(TU['target']=='UniRef90_T4BVE4')&(TU['target']=='UniRef90_D4IN85')&(TU['target']=='UniRef90_Q97TA2')&(TU['target']=='UniRef90_P0C0G7')]
TD[(TD['target']=='UniRef90_T4BVE4')&(TD['target']=='UniRef90_D4IN85')&(TD['target']=='UniRef90_Q97TA2')&(TD['target']=='UniRef90_P0C0G7')]
</code>
## networkx stuff
<code>
g=nx.from_pandas_edgelist(noHT[noHT[0]>10**-6].dropna(),source='source',target='target',edge_attr=0)
h=nx.from_pandas_edgelist(noHT[noHT[1]>10**-6].dropna(),source='source',target='target',edge_attr=1)
i=nx.from_pandas_edgelist(noHT[noHT[2]>10**-6].dropna(),source='source',target='target',edge_attr=2)
j=nx.from_pandas_edgelist(noHT[noHT[3]>10**-6].dropna(),source='source',target='target',edge_attr=3)
k=nx.from_pandas_edgelist(noHT[noHT[4]>10**-6].dropna(),source='source',target='target',edge_attr=4)
l=nx.from_pandas_edgelist(noHT[noHT[5]>10**-6].dropna(),source='source',target='target',edge_attr=5)
m=nx.from_pandas_edgelist(noHT[noHT[6]>10**-6].dropna(),source='source',target='target',edge_attr=6)
gg=nx.from_pandas_edgelist(HT[HT[0]>10**-6].dropna(),source='source',target='target',edge_attr=0)
hh=nx.from_pandas_edgelist(HT[HT[1]>10**-6].dropna(),source='source',target='target',edge_attr=1)
ii=nx.from_pandas_edgelist(HT[HT[2]>10**-6].dropna(),source='source',target='target',edge_attr=2)
jj=nx.from_pandas_edgelist(HT[HT[3]>10**-6].dropna(),source='source',target='target',edge_attr=3)
kk=nx.from_pandas_edgelist(HT[HT[4]>10**-6].dropna(),source='source',target='target',edge_attr=4)
ll=nx.from_pandas_edgelist(HT[HT[5]>10**-6].dropna(),source='source',target='target',edge_attr=5)
mm=nx.from_pandas_edgelist(HT[HT[6]>10**-6].dropna(),source='source',target='target',edge_attr=6)
</code>
<code>
fig=plt.figure(figsize=(5,15))
ax = fig.add_subplot(projection='3d')
LayeredNetworkGraph([g, h, i,j,k,l,m], ax=ax, layout=nx.spring_layout)
ax.set_axis_off()
# ax.figsize(5,15)
# plt.show()
fig.set_size_inches(20, 20)
fig.savefig('noHT.png', dpi=100)
</code>
<code>
fig=plt.figure(figsize=(5,15))
ax = fig.add_subplot(projection='3d')
LayeredNetworkGraph([g, h, i,j,k,l,m], ax=ax, layout=nx.spring_layout)
ax.set_axis_off()
# ax.figsize(5,15)
# plt.show()
fig.set_size_inches(20, 20)
fig.savefig('HT.png', dpi=100)
</code>
## 7 groups histograms
<code>
cc
</code>
<code>
cc=HT.merge(pd.DataFrame(aa),right_index=True,left_index=True)
# cc.rename(columns={'0_x':'0','0_y':'all'},inplace=True)
cc=cc.melt(['source','target'])
plt.figure(figsize=(15,5))
sns.histplot(data=cc, x="value", hue="variable", log_scale=True, element="step", fill=False,
cumulative=True, stat="density", common_norm=False)
</code>
<code>
sns.histplot(data=cc, x="value", log_scale=True, element="step", fill=False,hue='variable')
</code>
<code>
dd=noHT.merge(pd.DataFrame(aa),right_index=True,left_index=True)
# cc.rename(columns={'0_x':'0','0_y':'all'},inplace=True)
dd=dd.melt(['source','target'])
plt.figure(figsize=(15,5))
sns.histplot(
data=cc, x="value", hue="variable",
# hue_order=["Radial Velocity", "Transit"],
log_scale=True, element="step", fill=False,
cumulative=True, stat="density", common_norm=False,
)
</code>
<code>
sns.histplot(data=dd, x="value", log_scale=True, element="step", fill=False,hue='variable')
</code>
<code>
# tmp=plt.hist(HT.melt().value.dropna(how='all'),bins=100,log=True)
tmp=plt.hist(bb.values,bins=100,log=True)
tmp=plt.hist(aa.values,bins=100,log=True,alpha=.5)
</code>
## time plot
<code>
group_time_plot(noHT,7,10,'0')
</code>
<code>
jj['variable']=jj['variable'].astype('int')+2000
jj['variable'] = pd.to_datetime(jj['variable'], format='%Y')
</code>
<code>
# dd=(noHT-HT).dropna().melt()
# plt.figure(figsize=(15,5))
# sns.histplot(
# data=dd, x="value", hue="variable",
# # hue_order=["Radial Velocity", "Transit"],
# log_scale=True, element="step", fill=False,
# cumulative=True, stat="density", common_norm=False,
# )
jj=noHT.reset_index().melt(id_vars=['source','target'])
g = graphistry.edges(jj, 'source', 'target','value','variable')
g.plot()
</code>
<code>
group_time_plot(HT,7,10,'0')
</code>
<code>
# jj=jj.dropna(how='any')
# kk=kk.dropna(how='any')
# ll=jj.merge(kk,on=['source','target','time'],how='inner')
ll['vx']=rankdata(ll.value_x,method='min')
ll['vy']=rankdata(ll.value_y,method='min')
ll['diff']=ll['value_x']-ll['value_y']
ll['rank_diff']=ll['vx'].astype('int')-ll['vy'].astype('int')
ll['rank_diff']=np.abs(ll['rank_diff'])
ll.sort_values(by='rank_diff')
ll['species']=ll['source'].str.split('_').str[2]
# ll=ll[np.abs(ll['diff'])>1]
g = graphistry.edges(ll.dropna(how='any'), 'species', 'target','rank_diff')
g.plot()
</code>
## full time steps
<code>
# dd = dd/np.max(dd,axis=0)
dd=dd/np.sum(dd,axis=0)
### dd=np.argsort(dd)
noHT=dd.filter(regex='_0_').dropna(how='all')
HT1=dd.filter(regex='_1_')#.dropna(how='all')
HT2=dd.filter(regex='_2_')#.dropna(how='all')
HT=HT1.merge(HT2,right_index=True,left_index=True).dropna(how='all')
# [noHT.shape,HT1.shape,HT2.shape]
[noHT.shape,HT.shape]
jj=noHT.reset_index().melt(id_vars=['source','target'])
noHT.columns=noHT.columns.str.split('_').str[0]
# noHT.columns=pd.qcut((noHT.columns).astype('int'), 7, labels=False)
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT=noHT.dropna(how='all')
HT.columns=HT.columns.str.split('_').str[0]
# HT.columns=pd.qcut((HT.columns).astype('int'), 7, labels=False)
HT=HT.groupby(by=HT.columns, axis=1).mean()
HT=HT.dropna(how='all')
</code>
<code>
time_bar(jj,5,'rank','Anaerostipes')
# time_bar(jj,5,'value','all')
# time_bar(kk,5,'rank','all')
# time_bar(kk,5,'value','all')
# time_bar(ll,5,'rank_diff','all')
# time_bar(ll,5,'diff','all')
</code>
<code>
plt.hist(jj.value,log=True)
</code>
<code>
XX=10
labels,levels=pd.factorize(jj['target'])
jj['prot_id']=labels
jj['t']=jj['time'].astype('str').str.split('-').str[0]
labels,levels=pd.factorize(jj['t'])
jj['t']=labels
data=jj[['species','prot_id','time','t','value']]
df=data.copy()
grouped = data.groupby('prot_id')
def get_next_clus(x): return x['species'].shift(-1)
data["next_clus"] = grouped.apply(
lambda x: get_next_clus(x)).reset_index(0, drop=True)
# df = data[['species','target','t','value']]#.sort_values(['time'], ascending=[True]).groupby(['species','time']).max(5)
jeff=pd.DataFrame(df.groupby(['species','t'])['value'].nlargest(XX))
jeff.reset_index(inplace=True)
jeffA=data.loc[jeff['level_2']]
</code>
<code>
grouped = pd.DataFrame(jeffA.groupby(['species', 't'])['prot_id'].apply(list).groupby(level=0).apply(list))
jeff=grouped.explode('prot_id')
jeff.reset_index(inplace=True)
</code>
<code>
np.unique(data.species)
</code>
<code>
jeff2=jeff[jeff['species']=='Agathobaculum'].explode('prot_id')
# jeff2['prot_id']#.str.split(',')
jeff3=pd.DataFrame(jeff2['prot_id'].to_numpy().reshape(int(len(jeff2)/21),21))
# jeff3.index='Agathobaculum'
</code>
<code>
plt.hist((np.sort(jeff3.melt()['value'])).astype('str'),bins=50)#.value_counts())
</code>
<code>
plt.bar(x=np.arange(len((np.unique(jeff3.melt()['value'])))),height=jeff3.melt()['value'].value_counts()/21)
</code>
## crazy time plot & stats
run from gcn_plot.py script
<code>
import statsmodels.api as sm
from statsmodels.formula.api import ols
cw_lm=ols('value ~ C(variable)', data=cc[cc['variable']!='all']).fit() #Specify C for Categorical
print(sm.stats.anova_lm(cw_lm, typ=2))
</code>
<code>
import statsmodels.api as sm
from statsmodels.formula.api import ols
cw_lm=ols('value ~ C(variable)', data=dd[dd['variable']!='all']).fit() #Specify C for Categorical
print(sm.stats.anova_lm(cw_lm, typ=2))
</code>
## Sankey start
[from nicolasesnis github](https://gist.github.com/nicolasesnis/595d34c3c7dbca2b3419332304954433)
<code>
noHT=dd.filter(regex='_0_').dropna(how='all')
noHT.columns=noHT.columns.str.split('_').str[0]
# noHT.columns=pd.qcut((noHT.columns).astype('int'), 7, labels=False)
noHT=noHT.groupby(by=noHT.columns, axis=1).mean()
noHT.reset_index(inplace=True)
jj=noHT.melt(id_vars=['source','target'])
jj=jj.dropna(how='any')
jj.rename(columns={'variable':'t'},inplace=True)
# jj['t']=jj['variable'].astype('str').str.split('-').str[0]
labels,levels=pd.factorize(jj['t'])
jj['t']=labels
jj['species']=jj['source'].str.split('_').str[2]
data=jj[['species','target','t','value']]
df=data.copy()
# grouped = data.groupby('target')
XX=10
def get_next_clus(x): return x['species'].shift(-1)
data["next_clus"] = grouped.apply(
lambda x: get_next_clus(x)).reset_index(0, drop=True)
# df = data[['species','target','t','value']]#.sort_values(['time'], ascending=[True]).groupby(['species','time']).max(5)
jeff=pd.DataFrame(df.groupby(['species','t'])['value'].nlargest(XX))
jeff.reset_index(inplace=True)
jeffA=data.loc[jeff['level_2']]
grouped = jeffA.groupby('target')
def get_next_clus(x): return x['species'].shift(-1)
jeffA["next_clus"] = grouped.apply(
lambda x: get_next_clus(x)).reset_index(0, drop=True)
df = jeffA[['species','target','t','value']]#.sort_values(['time'], ascending=[True]).groupby(['species','time']).max(5)
jeffA.rename(columns={'target':'prot_id'},inplace=True)
data=jeffA
</code>
<code>
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts, meas, plotRidge,time_bar,group_time_plot,LayeredNetworkGraph,plot_sankey
labels,color,sources,targets,values=plot_sankey(data,XX,'prot_id','species')
print([len(targets),len(sources),len(values)])
# label = ["{} {}".format(node1_name, node1_val), "{} {}".format(node2_name, node2_val) ...]
fig = go.Figure(data=[go.Sankey(
node=dict(
thickness=15, # default is 20
line=dict(color="black", width=0.5),
label=labels,
color=colors
),
link=dict(
source=sources,
target=targets,
value=values,
# hovertemplate='%{link.label}.<br />',
# hovertemplate='%{value} top10 proteins went from %{source.label} to %{target.label}.<br />',
))])
fig.update_layout(autosize=True, title=dict(text="Evolution of Top "+str(XX)+" Proteins per Species", font_size=16),
font=dict(size=1, family="Arial"), plot_bgcolor='white')
# publish_to_web = True
# if publish_to_web:
py.iplot(fig, filename='prot_per_spec_evol')
# else:
# fig.show(renderer='chrome')
</code>
<code>
# noHT=dd.filter(regex='_0_').dropna(how='all')
HT.columns=HT.columns.str.split('_').str[0]
# noHT.columns=pd.qcut((noHT.columns).astype('int'), 7, labels=False)
HT=HT.groupby(by=HT.columns, axis=1).mean()
HT.reset_index(inplace=True)
jj=HT.melt(id_vars=['source','target'])
jj=jj.dropna(how='any')
jj.rename(columns={'variable':'t'},inplace=True)
# jj['t']=jj['variable'].astype('str').str.split('-').str[0]
labels,levels=pd.factorize(jj['t'])
jj['t']=labels
jj['species']=jj['source'].str.split('_').str[2]
data=jj[['species','target','t','value']]
df=data.copy()
# grouped = data.groupby('target')
XX=10
def get_next_clus(x): return x['species'].shift(-1)
data["next_clus"] = grouped.apply(
lambda x: get_next_clus(x)).reset_index(0, drop=True)
# df = data[['species','target','t','value']]#.sort_values(['time'], ascending=[True]).groupby(['species','time']).max(5)
jeff=pd.DataFrame(df.groupby(['species','t'])['value'].nlargest(XX))
jeff.reset_index(inplace=True)
jeffA=data.loc[jeff['level_2']]
grouped = jeffA.groupby('target')
def get_next_clus(x): return x['species'].shift(-1)
jeffA["next_clus"] = grouped.apply(
lambda x: get_next_clus(x)).reset_index(0, drop=True)
df = jeffA[['species','target','t','value']]#.sort_values(['time'], ascending=[True]).groupby(['species','time']).max(5)
jeffA.rename(columns={'target':'prot_id'},inplace=True)
data=jeffA
</code>
<code>
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts, meas, plotRidge,time_bar,group_time_plot,LayeredNetworkGraph,plot_sankey
labels,color,sources,targets,values=plot_sankey(data,XX,'prot_id','species')
print([len(targets),len(sources),len(values)])
# label = ["{} {}".format(node1_name, node1_val), "{} {}".format(node2_name, node2_val) ...]
fig = go.Figure(data=[go.Sankey(
node=dict(
thickness=15, # default is 20
line=dict(color="black", width=0.5),
label=labels,
color=colors
),
link=dict(
source=sources,
target=targets,
value=values,
# hovertemplate='%{link.label}.<br />',
# hovertemplate='%{value} top10 proteins went from %{source.label} to %{target.label}.<br />',
))])
fig.update_layout(autosize=True, title=dict(text="Evolution of Top "+str(XX)+" Proteins per Species", font_size=16),
font=dict(size=1, family="Arial"), plot_bgcolor='white')
# publish_to_web = True
# if publish_to_web:
py.iplot(fig, filename='prot_per_spec_evol')
# else:
# fig.show(renderer='chrome')
</code>
## try sankey again
<code>
# del data['next_clus']
data['uni_id']=data['species']+'_'+data['prot_id']
</code>
<code>
pd.to_datetime(data['t']+2000, format='%Y')
</code>
<code>
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts, meas, plotRidge,time_bar,group_time_plot,LayeredNetworkGraph,plot_sankey
labels,color,sources,targets,values=plot_sankey(data,XX,'prot_id','species')
print([len(targets),len(sources),len(values)])
# label = ["{} {}".format(node1_name, node1_val), "{} {}".format(node2_name, node2_val) ...]
fig = go.Figure(data=[go.Sankey(
node=dict(
thickness=15, # default is 20
line=dict(color="black", width=0.5),
label=labels,
color=colors
),
link=dict(
source=sources,
target=targets,
value=values,
# hovertemplate='%{link.label}.<br />',
# hovertemplate='%{value} top10 proteins went from %{source.label} to %{target.label}.<br />',
))])
fig.update_layout(autosize=True, title=dict(text="Evolution of Top "+str(XX)+" Proteins per Species", font_size=16),
font=dict(size=1, family="Arial"), plot_bgcolor='white')
# publish_to_web = True
# if publish_to_web:
py.iplot(fig, filename='prot_per_spec_evol')
# else:
# fig.show(renderer='chrome')
</code>
## network properties
<code>
# for net in dd:
# df=meas(measur,uni_bact,relgene,graphs)
ee=['nx.degree_centrality','nx.betweenness_centrality','nx.closeness_centrality','nx.node_redundancy','nx.communicability']
df=[meas(measur,uni_bact,relgene,graphs,patt) for measur in ee]
# df=[print(measur) for measur in dd]
</code>
<code>
measur='nx.degree'
HTXX=uni_bact[uni_bact.index.isin(relgene.columns[1:-2].str.split('-').str[0])]
HTXX['index']=np.arange(len(HTXX))
measur=eval(measur)
S = [measur(graphs[i]) for i in HTXX[HTXX['HT']==0]['index'].values[0:3]]
T = [measur(graphs[i]) for i in HTXX[HTXX['HT']!=0]['index'].values[0:3]]
if measur!=nx.degree:
non=pd.DataFrame(S).melt()
yes=pd.DataFrame(T).melt()
elif measur==nx.degree:
non=pd.DataFrame(S.pop())
non=non.rename(columns={0:'variable',1:'value'})
yes=pd.DataFrame(T.pop())
yes=yes.rename(columns={0:'variable',1:'value'})
# non=pd.DataFrame(S).melt()
non['type']='NoHT'
non.dropna(inplace=True)
non=non[non.value!=0]
non=non[~non['variable'].str.contains('UniRef90')]
non.value=non.value/np.sum(non.value)
yes['type']='HT'
yes.dropna(inplace=True)
yes=yes[yes.value!=0]
yes=yes[~yes['variable'].str.contains('UniRef90')]
yes.value=yes.value/np.sum(yes.value)
df=non.append(yes)
df=df.dropna()
df['gen']=df.variable.str.split('_').str[2]
</code>
# analyze ARG via networkX
<code>
from networkx import bipartite
from functools import reduce
def bipart(G,method,nodes):
return pd.DataFrame.from_dict(eval('bipartite.'+method)(G,nodes),orient='index',columns=[str(method)])
patt='all'
sys.path.insert(1, './run/gcn/')
import gcn_func
importlib.reload(sys.modules['gcn_func'])
from gcn_func import pdize_net, plot_comm, load_list_of_dicts
</code>
<code>
# patt='all'
sys.path.insert(1, './run/gcn/')
import gcn_func
importlib.reload(sys.modules['gcn_func'])
from gcn_func import bip, load_list_of_dicts,calc_bipart, meas, time_bar,proc_dat,rev_tbar,group_time_plot,time_order_net,build_gcn,shuffle_net,structural_analysis
relgene=pd.read_csv('all_arg_subset_genefamilies-cpm.tsv',sep='\t',nrows=10)
Ngraphs = load_list_of_dicts('data/gcn/NX_Emore_ARG.pkl')
Bgraphs = load_list_of_dicts('data/gcn/BXX_Emore_ARG.pkl')
B2graphs = load_list_of_dicts('data/gcn/B2X_Emore_ARG.pkl')
# relgene=pd.read_csv('all_arg_subset_genefamilies-cpm.tsv',sep='\t',nrows=1)
# graphs = load_list_of_dicts('data/gcn/NX_Emore_ARG.pkl')
ARG_meta=pd.read_excel('run/gcn/ARG_treatment_infor_modified.xlsx',index_col=0)
ARG_meta2=pd.read_excel('run/gcn/patients_Tx_batch3_for_DM.xlsx',index_col=None,skiprows=1,names=['id','group'])
relgene.columns=relgene.columns.str.replace("-00", "-00ST")
relgene.columns=relgene.columns.str.replace("-00STST", "-00ST")
relgene.columns=relgene.columns.str.split('-').str[0]+'-'+relgene.columns.str.split('-').str[1]
ARG_meta['id']=ARG_meta['id'].str.replace('-00ST','')
META=pd.concat([pd.DataFrame(ARG_meta[['id','group']]),ARG_meta2],ignore_index=True)
</code>
<code>
OTHER_00ST = load_list_of_dicts('data/gcn/OTHER_00ST.pkl')
OTHER_01ST = load_list_of_dicts('data/gcn/OTHER_01ST.pkl')
OTHER_02ST = load_list_of_dicts('data/gcn/OTHER_02ST.pkl')
CLA_00ST = load_list_of_dicts('data/gcn/CLA_00ST.pkl')
CLA_01ST = load_list_of_dicts('data/gcn/CLA_01ST.pkl')
CLA_02ST = load_list_of_dicts('data/gcn/CLA_02ST.pkl')
LEVO_00ST = load_list_of_dicts('data/gcn/LEVO_00ST.pkl')
LEVO_01ST = load_list_of_dicts('data/gcn/LEVO_01ST.pkl')
LEVO_02ST = load_list_of_dicts('data/gcn/LEVO_02ST.pkl')
</code>
<code>
O_0b,O_0c,O_0d=pdize_net(OTHER_00ST)
O_1b,O_1c,O_1d=pdize_net(OTHER_01ST)
O_2b,O_2c,O_2d=pdize_net(OTHER_02ST)
C_0b,C_0c,C_0d=pdize_net(CLA_00ST)
C_1b,C_1c,C_1d=pdize_net(CLA_01ST)
C_2b,C_2c,C_2d=pdize_net(CLA_02ST)
L_0b,L_0c,L_0d=pdize_net(LEVO_00ST)
L_1b,L_1c,L_1d=pdize_net(LEVO_01ST)
L_2b,L_2c,L_2d=pdize_net(LEVO_02ST)
# for j in ['C_0b','C_1b','C_2b','L_0b','L_1b','L_2b','O_0b','O_1b','O_2b']:
# j=eval(j).reset_index()
# j['source']=j['source'].str.split('_').str[2]
# print(len(j.groupby('source').count()))
</code>
## antibiotic annotation CARDS
via [uniprot](https://www.uniprot.org/uploadlists/) queries [here](https://www.uniprot.org/uniprot/?query=yourlist:M20220503F248CABF64506F29A91F8037F07B67D14C0F834&sort=yourlist:M20220503F248CABF64506F29A91F8037F07B67D14C0F834&columns=yourlist(M20220503F248CABF64506F29A91F8037F07B67D14C0F834),id,entry%20name,reviewed,protein%20names,genes,organism,length)
and [via](https://www.uniprot.org/uniprot/?query=yourlist:M20220503A084FC58F6BBA219896F365D15F2EB444BBF42C&sort=yourlist:M20220503A084FC58F6BBA219896F365D15F2EB444BBF42C&columns=yourlist(M20220503A084FC58F6BBA219896F365D15F2EB444BBF42C),id,entry%20name,reviewed,protein%20names,genes,organism,length)
<code>
uni_conv=pd.read_csv('run/gcn/CLA_uniref_conv_full.txt',sep='\t')
# card=pd.read_csv('run/gcn/card_out/aro_index.tsv',sep='\t')
# card['Protein Accession'].to_csv('card_prot.txt',sep='\t')
card=pd.read_csv('run/gcn/card_prot_conv.txt',sep='\t')
def card_net(OTHER_00ST,uni_conv,card):
bb=pd.DataFrame(columns=['source','target'])
for i,ii in enumerate(OTHER_00ST):
cc=nx.to_pandas_edgelist(OTHER_00ST[i])
cc['weight']=1
bb=bb.merge(cc,how='outer',right_on=['source','target'],left_on=['source','target'])
bb.fillna(0)
# aa=pd.DataFrame(bb.set_index(['source','target']).fillna(0).mean(axis=1))
# ee=aa
bb.reset_index(inplace=True)
cc=bb.loc[:,bb.columns.str.contains('weight')]
cc=1-(np.sum(cc,axis=0)/len(cc))
bb['target']=bb['target'].str.split('_').str[1]#.to_csv('CLA_uniref.txt',sep='\t')
bb=bb.merge(uni_conv,left_on='target',right_on='Entry')
bb=bb.merge(card,on='Entry name')#[['source','target',0]]
bb.fillna(0,inplace=True)
ee=bb.loc[:,bb.columns.str.contains('weight')]
dd=1-(np.sum(ee,axis=0)/len(ee))
return bb,cc,dd
</code>
<code>
OTHER__00ST,OTHER__00ST_full,OTHER__00ST_card=card_net(OTHER_00ST,uni_conv,card)
OTHER__01ST,OTHER__01ST_full,OTHER__01ST_card=card_net(OTHER_01ST,uni_conv,card)
OTHER__02ST,OTHER__02ST_full,OTHER__02ST_card=card_net(OTHER_02ST,uni_conv,card)
CLA__00ST,CLA__00ST_full,CLA__00ST_card=card_net(CLA_00ST,uni_conv,card)
CLA__01ST,CLA__01ST_full,CLA__01ST_card=card_net(CLA_01ST,uni_conv,card)
CLA__02ST,CLA__02ST_full,CLA__02ST_card=card_net(CLA_02ST,uni_conv,card)
LEVO__00ST,LEVO__00ST_full,LEVO__00ST_card=card_net(LEVO_00ST,uni_conv,card)
LEVO__01ST,LEVO__01ST_full,LEVO__01ST_card=card_net(LEVO_01ST,uni_conv,card)
LEVO__02ST,LEVO__02ST_full,LEVO__02ST_card=card_net(LEVO_02ST,uni_conv,card)
</code>
<code>
C_00=CLA__00ST.loc[:,CLA__00ST.columns.str.contains('weight')]
C_00[['source','target']]=CLA__00ST[['Organism_x','Gene names_x']]
# O_00.set_index(['source','target'],inplace=True)
C_00
</code>
<code>
C_00['target'].tolist()
</code>
<code>
[sum(c+0),sum(d+0),sum(e+0)]
</code>
<code>
sparsity=pd.DataFrame()
for j,ii in enumerate(['OTHER__00ST','OTHER__01ST','OTHER__02ST','CLA__00ST','CLA__01ST','CLA__02ST','LEVO__00ST','LEVO__01ST','LEVO__02ST']):
for p in (['full','card']):
c=pd.DataFrame(eval(ii+'_'+p))
c['trx']=ii.split('__')[0]
c['time']=ii.split('__')[1]
c['size']=p
sparsity=sparsity.append(c)
sparsity.rename(columns={0:'sparsity'},inplace=True)
</code>
<code>
sparsity=sparsity[sparsity['sparsity']!=1]
g = sns.FacetGrid(sparsity,col='trx',row='size')#,col_order=ordered_days)#,row='metric')
### change this to 4 ^
g.map(sns.boxplot,'time','sparsity',palette='muted',order=['00ST','01ST','02ST'])
g.map(sns.swarmplot,'time','sparsity',order=['00ST','01ST','02ST'],color=".25")
box_pairs=[(('00ST'),('01ST')),(('01ST'),('02ST')),(('00ST'),('02ST'))]
for ax in g.axes.flatten():
ax.tick_params(labelbottom=True)
trxx=ax.get_title().split('|')[1].split('=')[1][1:]
mm=ax.get_title().split('|')[0].split('=')[1][1:-1]
data=sparsity[(sparsity['trx']==str(trxx))&(sparsity['size']==str(mm))]
annotator =Annotator(ax, data=data, x='time', y='sparsity',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind', text_format='simple', loc='inside',comparisons_correction='fdr_bh')
annotator.apply_and_annotate()
plt.tight_layout()
plt.show()
</code>
<code>
sparsity
</code>
## time
<code>
# np.round(LL.value,0)
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('source').sum()
LL.columns=['00ST','01ST','02ST']
LL=LL[((np.round(LL['00ST'],0))==(np.round(LL['01ST'],0)))&((np.round(LL['02ST'],0))==(np.round(LL['01ST'],0)))]
LL=LL.reset_index().melt('source')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('source').sum()
LL.columns=['00ST','01ST','02ST']
# LL=LL[(LL['00ST']==0)&(LL['01ST']!=0)&(LL['02ST']==0)]
LL=LL[(LL['00ST']<LL['01ST']/2)&(LL['01ST']/2>LL['02ST'])]
LL=LL.reset_index().melt('source')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('source').sum()
LL.columns=['00ST','01ST','02ST']
# LL=LL[(LL['00ST']!=0)&(LL['01ST']==0)&(LL['02ST']!=0)]
LL=LL[(LL['00ST']>LL['01ST']*2)&(LL['01ST']*2<LL['02ST'])]
LL=LL.reset_index().melt('source')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('source').sum()
LL.columns=['00ST','01ST','02ST']
LL=LL[(LL['00ST']>LL['01ST'])&(LL['01ST']>LL['02ST'])]
# CLA_val=FRcalc[(FRcalc['00']>FRcalc['01'])&(FRcalc['01']<FRcalc['02'])]
LL=LL.reset_index().melt('source')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('source').sum()
LL.columns=['00ST','01ST','02ST']
LL=LL[(LL['00ST']<LL['01ST'])&(LL['01ST']<LL['02ST'])]
LL=LL.reset_index().melt('source')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL=LL.groupby('target').sum()
LL.columns=['00ST','01ST','02ST']
# LL=LL[(LL['00ST']==0)&(LL['01ST']!=0)&(LL['02ST']==0)]
LL=LL[(LL['00ST']<LL['01ST']/5)&(LL['01ST']/5>LL['02ST'])]
LL=LL.reset_index().melt('target')
LL.source=LL.source.str.split('_').str[2]
sns.relplot(data=LL, x="variable", y=(LL['value']),hue="source",kind="line")
</code>
<code>
def get_graph_info(graph):
print("Number of nodes:", graph.number_of_nodes())
print("Number of edges:", graph.number_of_edges())
print("Average Cluster Coefficients:", nx.average_clustering(graph))
print("Connected components:", len(list(nx.connected_components(graph))))
</code>
<code>
LL=(L_0b).merge(L_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(L_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL.columns=['00','01','02']
LL=LL.reset_index()#.melt(['source','target'])
# LL['variable']=LL['variable'].astype('int')+2000
# LL['variable'] = pd.to_datetime(LL['variable'], format='%Y')
# LL=LL[LL.value>0]
# graphistry.edges(LL, 'source', 'target','value','variable').plot()
</code>
<code>
# M.reset_index(inplace=True)
# M[np.argsort(np.sum(M,axis=1)),:]
LL
</code>
<code>
M=LL.pivot('source',columns='target',values='00').fillna(0)
# N=M[np.argsort(np.sum(M,axis=1)),:][:,np.argsort(np.sum(M,axis=0))]
# plt.imshow(N, cmap='hot', interpolation='nearest',aspect='auto')
# plt.show()
</code>
<code>
AA=LL.pivot('source',columns='target',values='01').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
AA=LL.pivot('source',columns='target',values='02').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
OTHERc=O_0c.merge(O_1c,on=['target']).merge(O_2c,on=['target'])
OTHERc.columns=['00ST','01ST','02ST']
CLAc=C_0c.merge(C_1c,on=['target']).merge(C_2c,on=['target'])
CLAc.columns=['00ST','01ST','02ST']
LEVOc=L_0c.merge(L_1c,on=['target']).merge(L_2c,on=['target'])
LEVOc.columns=['00ST','01ST','02ST']
</code>
<code>
OTHERd=O_0d.merge(O_1d,on=['source']).merge(O_2d,on=['source'])
OTHERd.columns=['00ST','01ST','02ST']
CLAd=C_0d.merge(C_1d,on=['source']).merge(C_2d,on=['source'])
CLAd.columns=['00ST','01ST','02ST']
LEVOd=L_0d.merge(L_1d,on=['source']).merge(L_2d,on=['source'])
LEVOd.columns=['00ST','01ST','02ST']
</code>
<code>
# tmp=plt.hist(O_0,log=True,bins=100)
# tmp=plt.hist(O_1,log=True,bins=100)
# tmp=plt.hist(O_2,log=True,bins=100)
sns.scatterplot(data=OTHERc, x="00ST", y="01ST")
sns.scatterplot(data=OTHERc, x="01ST", y="02ST")
plt.plot([0, 14], [0, 14],ls='--',color='grey')
# sns.scatterplot(data=OTHER, x="00ST", y="02ST")
</code>
<code>
sns.scatterplot(data=OTHERd, x="00ST", y="01ST")
sns.scatterplot(data=OTHERd, x="01ST", y="02ST")
plt.plot([0, 800], [0, 800],ls='--',color='grey')
</code>
<code>
# tmp=plt.hist(np.mean(C_0.groupby('target').sum(),axis=1),log=True,bins=100)
# tmp=plt.hist(np.mean(C_1.groupby('target').sum(),axis=1),log=True,bins=100)
# tmp=plt.hist(np.mean(C_2.groupby('target').sum(),axis=1),log=True,bins=100)
sns.scatterplot(data=CLAc, x="00ST", y="01ST")
sns.scatterplot(data=CLAc, x="01ST", y="02ST")
# sns.scatterplot(data=CLA, x="00ST", y="02ST")
plt.plot([0, 14], [0, 14],ls='--',color='grey')
</code>
<code>
sns.scatterplot(data=CLAd, x="00ST", y="01ST")
sns.scatterplot(data=CLAd, x="01ST", y="02ST")
plt.plot([0, 800], [0, 800],ls='--',color='grey')
</code>
<code>
# tmp=plt.hist(np.mean(L_0.groupby('target').sum(),axis=1),log=True,bins=100)
# tmp=plt.hist(np.mean(L_1.groupby('target').sum(),axis=1),log=True,bins=100)
# tmp=plt.hist(np.mean(L_2.groupby('target').sum(),axis=1),log=True,bins=100)
sns.scatterplot(data=LEVOc, x="00ST", y="01ST")
sns.scatterplot(data=LEVOc, x="01ST", y="02ST")
# sns.scatterplot(data=LEVO, x="01ST", y="02ST")
plt.plot([0, 14], [0, 14],ls='--',color='grey')
</code>
<code>
sns.scatterplot(data=LEVOd, x="00ST", y="01ST")
sns.scatterplot(data=LEVOd, x="01ST", y="02ST")
plt.plot([0, 800], [0, 800],ls='--',color='grey')
</code>
## tracking temporal communities
[example code here](https://stackoverflow.com/questions/69064622/igraph-plot-for-multiple-layers-partition-does-not-return-expected-output)
<code>
LL=(C_0b).merge(C_1b,right_on=['source','target'],left_on=['source','target'],how='outer').merge(C_2b,right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL.columns=['00ST','01ST','02ST']
</code>
<code>
# G=nx.from_pandas_edgelist(LL['00ST'].reset_index(),source='source',target='target',edge_attr='00ST')
# G_0 = ig.Graph.from_networkx(G)
# # G_1.vs['id']=G.nodes
# G=nx.from_pandas_edgelist(LL['01ST'].reset_index(),source='source',target='target',edge_attr='01ST')
# G_1 = ig.Graph.from_networkx(G)
# # G_2.vs['id']=G.nodes
# G=nx.from_pandas_edgelist(LL['02ST'].reset_index(),source='source',target='target',edge_attr='02ST')
# G_2 = ig.Graph.from_networkx(G)
# # G_3.vs['id']=G.nodes
# # G_1.vs['id'] = G_1.nodes
# print([G_0.is_bipartite(),G_1.is_bipartite(),G_2.is_bipartite()])
# # G_2 = ig.Graph.from_networkx(G_2)
# # G_3 = ig.Graph.from_networkx(G_3)
</code>
<code>
# G_1 = ig.Graph.DataFrame(LL['00ST'].reset_index())
# G_2 = ig.Graph.DataFrame(LL['01ST'].reset_index())
# G_3 = ig.Graph.DataFrame(LL['02ST'].reset_index())
jj0=pd.DataFrame(LL['00ST'].reset_index())
jj0.columns=['start','end','value']
jj0=jj0[jj0['value']>0]
kk=pd.DataFrame(np.unique(jj0[['start','end']].melt()['value']))
kk.columns=['name']
kk['id']=kk['name']
kk['types']=np.concatenate([np.ones(len(np.unique(jj0.end))),np.zeros(len(np.unique(jj0.start)))]).astype('int')
G_00=igraph_from_pandas(edges_table=jj0, vertices_table=kk, source_cl='start', target_cl='end', vertex_attrs=list(kk.columns), vertex_id_cl='name', directed=False)
jj1=pd.DataFrame(LL['01ST'].reset_index())
jj1.columns=['start','end','value']
jj1=jj1[jj1['value']>0]
kk=pd.DataFrame(np.unique(jj1[['start','end']].melt()['value']))
kk.columns=['name']
kk['id']=kk['name']
kk['types']=np.concatenate([np.ones(len(np.unique(jj1.end))),np.zeros(len(np.unique(jj1.start)))]).astype('int')
G_11=igraph_from_pandas(edges_table=jj1, vertices_table=kk, source_cl='start', target_cl='end', vertex_attrs=list(kk.columns), vertex_id_cl='name', directed=False)
jj2=pd.DataFrame(LL['02ST'].reset_index())
jj2.columns=['start','end','value']
jj2=jj2[jj2['value']>0]
kk=pd.DataFrame(np.unique(jj2[['start','end']].melt()['value']))
kk.columns=['name']
kk['id']=kk['name']
kk['types']=np.concatenate([np.ones(len(np.unique(jj2.end))),np.zeros(len(np.unique(jj2.start)))]).astype('int')
G_22=igraph_from_pandas(edges_table=jj2, vertices_table=kk, source_cl='start', target_cl='end',vertex_attrs=list(kk.columns), vertex_id_cl='name', directed=False)
print([G_11.is_bipartite(),G_22.is_bipartite(),G_00.is_bipartite()])
</code>
## [community detection](https://igraph.org/c/doc/igraph-Community.html#igraph_community_multilevel)
<code>
def ready_net(C):#C in ['C','L','O']:
LL=eval(str(C)+'_0b').merge(eval(str(C)+'_1b'),right_on=['source','target'],left_on=['source','target'],how='outer').merge(eval(str(C)+'_2b'),right_on=['source','target'],left_on=['source','target'],how='outer').fillna(0)
LL.columns=['00ST','01ST','02ST']
# modes = pd.concat([LL['mode'] for df in (df1, df2, df3, df4)], ignore_index=True).unique()
LL.reset_index(inplace=True)
LL['source']=LL['source'].str.split('_').str[2]
return LL
CLA=ready_net('C')
LEVO=ready_net('L')
OTHER=ready_net('O')
</code>
<code>
species=pd.unique(np.concatenate([CLA['source'],LEVO['source'],OTHER['source']]))
colors = sns.color_palette('hls', len(species))
palette = {mode: color for mode, color in zip(species, colors)}
</code>
<code>
def plot_comm(CC,LL,OO,palette,j): ##time, community_type
c=0
fig, ax = plt.subplots(ncols=3, nrows=3,figsize=(15,15))
# dict_keys = [k for k in z.keys()]
for k in [CC,LL,OO]:
web=(k)
k=eval(k)
for i in k.columns[2:]:
jj0=pd.DataFrame(k[['source','target',i]])#.reset_index()
jj0.columns=['start','end','value']
jj0=jj0[jj0['value']>0]
kk=pd.DataFrame(np.unique(jj0[['start','end']].melt()['value']))
kk.columns=['name']
kk['id']=kk['name']
kk['types']=np.concatenate([np.ones(len(np.unique(jj0.end))),np.zeros(len(np.unique(jj0.start)))]).astype('int')
G_00=igraph_from_pandas(edges_table=jj0, vertices_table=kk, source_cl='start', target_cl='end', vertex_attrs=list(kk.columns), vertex_id_cl='name', directed=False)
G_00.vs['pagerank']=G_00.pagerank()
G_00.vs['cluster'] = G_00.community_infomap().membership
# N,Q,I,R=Parallel(n_jobs=10)(structural_analysis(ii,i,graphs,ARG_meta,rand=rand,deg_rand=deg_rand))
# for j in [edge_betweenness,fastgreedy,infomap,label_propagation,leading_eigenvector,leiden,multilevel,_optimal_modularity,spinglass,walktrap]:
# G_00.vs['louvain_membership']=G_00.community_multilevel().membership
try:
Path("run/gcn/img/"+j).mkdir(parents=True, exist_ok=True)
G_00.vs[j+'membership']=eval('G_00.community_'+j)().membership
except:
G_00.vs[j+'membership']=eval('G_00.community_'+j)()
g00e=G_00.get_edge_dataframe()#.reset_index()
g00v=G_00.get_vertex_dataframe().reset_index()
g00e['weight']=jj0['value']
jj=g00e.merge(g00v,left_on='source',right_on='vertex ID').merge(g00v,left_on='target',right_on='vertex ID')
jj.name_x, jj.name_y = np.where(jj.name_x.str.contains('UniRef'), [jj.name_y, jj.name_x], [jj.name_x, jj.name_y])
# for j in ['louvain_','leiden_']:
gg=jj.groupby(['name_x',j+'membership_x']).count().sort_values(['target',j+'membership_x'])
gg=gg.reset_index()[['name_x',j+'membership_x','target']]
# hh=hh.merge(gg,how='outer')
# gg[j+'membership_x']=gg[j+'membership_x'].astype('category')
sns.histplot(gg, x=gg[j+'membership_x'].astype('int')+1, hue='name_x',log_scale=[False,True], weights='target',multiple='stack',shrink=0.8,palette=palette,ax=ax.flat[c],legend=False).set_title(web+"_"+i)
c=c+1
# plt.figure(figsize=(10,6))
# plt.set(xlabel=i+' '+j+' membership',yscale='log',ylabel='link count')
# g.set_title(str(C)+'_'+i+'_'+j)
# g.set_xlabel(i+' '+j+' membership')
# g.set_yscale('log')
plt.savefig('run/gcn/img/'+j+'.png', dpi=100)
</code>
<code>
for j in ['multilevel','label_propagation','spinglass','infomap','leiden']:
plot_comm('CLA','LEVO','OTHER',palette,j)
</code>
<code>
# partitions[0].graph
# partitions[0].graph.vs['pagerank']=partitions[0].graph.pagerank()
# partitions[0].graph.vs['cluster'] = partitions[0].graph.community_infomap().membership
# partitions[0].graph.vs['membership']=partitions[0].membership
# plotter = graphistry.bind(source='source', destination='target')
# plotter.bind(point_color='membership', point_size='pagerank').plot(partitions[0].graph)
# G22_comms.graph.vs['pagerank']=G22_comms.graph.pagerank()
# G22_comms.graph.vs['cluster'] = G22_comms.graph.community_infomap().membership
# G22_comms.graph.vs['membership']=G22_comms.membership
# import graphistry, igraph, pandas as pd
# g = graphistry.edges(g00e, source='source', destination='target')
# g2 = (g.nodes(g00v, 'vertex ID').bind(point_title='name').bind(edge_title='weight'))
# ig = g2.to_igraph(directed=False)
# ig.vs['cluster'] = ig.community_infomap().membership
g3a = graphistry.from_igraph(G_22, load_edges=True, load_nodes=True,node_attributes=[,'vertex ID', 'cluster'])
g3a._nodes.sample(3)
g3b = g3a.nodes(g3a._nodes.assign(color=g3a._nodes['cluster'].apply(lambda x: x % 9).astype('int32')))
g3 = g3b.encode_point_color('color').bind(edge_weight='counts')
g3.plot()
# load just the desired attributes: reuse original edges, and enrich with just cluster
# g3a = g2.from_igraph(ig, load_edges=False, node_attributes=['vertex ID', 'cluster'])
# g3a._nodes.sample(3)
# plotter = graphistry.bind(source='source', destination='target')
# plotter.bind( point_size='cluster').plot(G_00)
# G_00.vs['pagerank']=G_00.pagerank()
# G_11.vs['pagerank']=G_11.pagerank()
# G_22.vs['pagerank']=G_22.pagerank()
# G_00.vs['cluster'] = G_00.community_infomap().membership
# G_11.vs['cluster'] = G_11.community_infomap().membership
# G_22.vs['cluster'] = G_22.community_infomap().membership
</code>
## resume partition
<code>
optimiser = la.Optimiser()
G_coupling = ig.Graph.Formula('1 -- 2 -- 3');
G_coupling.es['weight'] = 0.1; # Interslice coupling strength
G_coupling.vs['slice'] = [G_00, G_11, G_22]
# G_coupling.vs['types'] = [G_11.vs['types'], G_22.vs['types'], G_33.vs['types']]
node_size=0
gamma=0
</code>
<code>
# membership, improvement = la.find_partition_multiplex([G_1, G_2, G_3],la.ModularityVertexPartition)
layers, interslice_layer, G_full = la.slices_to_layers(G_coupling);
partitions = [la.CPMVertexPartition(H, node_sizes='node_size',weights='weight',resolution_parameter=gamma)for H in layers]
partitions.append(la.CPMVertexPartition(interslice_layer, resolution_parameter=0,node_sizes='node_size', weights='weight'))
# diffA = optimiser.optimise_partition_multiplex(partitions + [interslice_partition])
</code>
<code>
# membership, improvement = la.find_partition_multiplex([G_1, G_2, G_3],la.ModularityVertexPartition)
layers, interslice_layer, G_full = la.time_slices_to_layers([G_00, G_11, G_22],interslice_weight=0.1)
partitions = [la.CPMVertexPartition(H, node_sizes='node_size',weights=1,resolution_parameter=.5)for H in layers]
partitions.append(la.CPMVertexPartition(interslice_layer, resolution_parameter=0,node_sizes='node_size', weights='weight'))
# diffB = optimiser.optimise_partition_multiplex(partitions + [interslice_partition])
</code>
<code>
# G0=partitions[2].graph.get_edge_dataframe().sort_values(by=['source','target'])
# AA=G0.pivot('source',columns='target',values='weight').fillna(0)
# plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
# plt.show()
# tmp=plt.hist(G0['source'],bins=100,log=True)
# tmp=plt.hist(G0['target'],bins=100,log=True,alpha=.5)
</code>
<code>
# df = partitions[2].graph.get_edge_dataframe()
# df_vert = partitions[2].graph.get_vertex_dataframe()
# df['source'].replace(df_vert['name'], inplace=True)
# df['target'].replace(df_vert['name'], inplace=True)
# df_vert.set_index('name', inplace=True) # Optional
# df_vert['membership']=partitions[1].membership
</code>
<code>
g = G_00.to_graph_tool()
in_hist = vertex_hist(g, "in")
y = in_hist[0]
err = sqrt(in_hist[0])
err[err >= y] = y[err >= y] - 1e-2
figure(figsize=(6,4))
errorbar(in_hist[1][:-1], in_hist[0], fmt="o", yerr=err,
label="00ST_in_deg")
g = G_11.to_graph_tool()
in_hist = vertex_hist(g, "in")
y = in_hist[0]
err = sqrt(in_hist[0])
err[err >= y] = y[err >= y] - 1e-2
# figure(figsize=(6,4))
errorbar(in_hist[1][:-1], in_hist[0], fmt="o", yerr=err,
label="01ST_in_deg")
g = G_22.to_graph_tool()
in_hist = vertex_hist(g, "in")
y = in_hist[0]
err = sqrt(in_hist[0])
err[err >= y] = y[err >= y] - 1e-2
# figure(figsize=(6,4))
errorbar(in_hist[1][:-1], in_hist[0], fmt="o", yerr=err,
label="02ST_in_deg")
gca().set_yscale("log")
gca().set_xscale("log")
gca().set_ylim(1e-1, 1e4)
gca().set_xlim(0.8, 1e2)
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
subplots_adjust(left=0.2, bottom=0.2)
xlabel("$k_{in}$")
ylabel("$NP(k_{in})$")
tight_layout()
</code>
<code>
state = gt.minimize_nested_blockmodel_dl(g)
</code>
<code>
state.draw()
</code>
<code>
state_ndc = gt.minimize_nested_blockmodel_dl(g, state_args=dict(deg_corr=False))
state_dc = gt.minimize_nested_blockmodel_dl(g, state_args=dict(deg_corr=True))
print("Non-degree-corrected DL:\t", state_ndc.entropy())
print("Degree-corrected DL:\t", state_dc.entropy())
</code>
<code>
print(u"ln \u039b: ", state_dc.entropy() - state_ndc.entropy())
</code>
<code>
state = gt.BlockState(g) # This automatically initializes the state with a partition
# into one group. The user could also pass a higher number
# to start with a random partition of a given size, or pass a
# specific initial partition using the 'b' parameter.
# Now we run 1,000 sweeps of the MCMC. Note that the number of groups
# is allowed to change, so it will eventually move from the initial
# value of B=1 to whatever is most appropriate for the data.
dS, nattempts, nmoves = state.multiflip_mcmc_sweep(niter=1000)
print("Change in description length:", dS)
print("Number of accepted vertex moves:", nmoves)
</code>
<code>
levels = state.get_levels()
for s in levels:
print(s)
if s.get_N() == 1:
break
</code>
<code>
tree = min_spanning_tree(g)
graph_draw(g, edge_color=tree,output="min_tree.svg"))
g.set_edge_filter(tree)
graph_draw(g,output="min_tree_filtered.svg")
</code>
<code>
g = G_11.to_graph_tool()
state = gt.minimize_blockmodel_dl(g, state=gt.PPBlockState)
state.multiflip_mcmc_sweep(beta=np.inf, niter=100)
# pos = sfdp_layout(g)
# state.draw(g)#, output_size=(1000, 1000), vertex_color=[1,1,1,0])
# vertex_size=1, edge_pen_width=1.2)#,
# vcmap=[plt].cm.gist_heat_r)
state.draw(output='')#pos=g.vp.pos)
</code>
<code>
# red_cm = plt.colors.LinearSegmentedColormap.from_list("Set3", clrs)
# draw red edge last
eorder = u.ep.eprob.copy()
eorder.a *= -1
bstate.draw(pos=u.own_property(g.vp.pos), vertex_shape="pie", vertex_pie_fractions=pv,
edge_pen_width=gt.prop_to_size(ew, .1, 4, power=1),
edge_gradient=None, edge_color=u.ep.eprob, #ecmap=red_cm,
eorder=eorder)
</code>
<code>
G0=interslice_layer.to_networkx()
G1=layers[0].to_networkx()#.to_pandas_adjacency()
G2=layers[1].to_networkx()#.to_pandas_adjacency()
G3=layers[2].to_networkx()#.to_pandas_adjacency()
</code>
<code>
G0=nx.to_pandas_edgelist(G0)
G0=G0[G0['weight']>0]
G1=nx.to_pandas_edgelist(G1)
G1=G1[G1['weight']>0]
G2=nx.to_pandas_edgelist(G2)
G2=G2[G2['weight']>0]
G3=nx.to_pandas_edgelist(G3)
G3=G3[G3['weight']>0]
# G1=nx.to_pandas_adjacency(G1)
# G2=nx.to_pandas_adjacency(G2)
# G3=nx.to_pandas_adjacency(G3)
# partitions
</code>
<code>
AA=G0.pivot('source',columns='target',values='weight').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
AA=G1.pivot('source',columns='target',values='weight').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
AA=G2.pivot('source',columns='target',values='weight').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
AA=G3.pivot('source',columns='target',values='weight').fillna(0)
plt.imshow(AA, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
fig=plt.figure(figsize=(5,15))
ax = fig.add_subplot(projection='3d')
LayeredNetworkGraph([G0,G1,G2], ax=ax, layout=nx.spring_layout)
ax.set_axis_off()
# ax.figsize(5,15)
# plt.show()
fig.set_size_inches(20, 20)
fig.savefig('LEVO_itemp_comm.png', dpi=100)
</code>
# summary network metrics
<code>
cc=pd.read_csv('~/data/gcn/comp_net/post_nest_clust_AA.txt',sep='\t',names=['name','AC','RAC','D','SB'])
AVG=cc.melt('name')
AVG['trx']=AVG['name'].str.split('_').str[1]
AVG['time']=AVG['name'].str.split('_').str[2]
AVG['metric']=AVG['variable']
</code>
<code>
# LCC=LC.melt()
# LCC['trx']=LCC.variable.str.split('_').str[1]
# LCC['time']=LCC.variable.str.split('_').str[2]
# LCC['metric']=LCC.variable.str.split('_').str[3]
# LCC=LCC[LCC.value!=0]
# LC.columns=AVG.columns.str.replace("_00", "_00ST")
# LC.columns=LC.columns.str.replace("_00STST", "_00ST")
AVG['time']=AVG['time'].replace("03ST", "02ST")
ordered_days = sorted(AVG['trx'].unique())
g = sns.FacetGrid(AVG,col='trx',col_order=ordered_days,row='metric')
# change this to 4 ^
g.map(sns.boxplot,'time','value',palette='muted')
box_pairs=[(('00ST'),('01ST')),(('01ST'),('02ST')),(('00ST'),('02ST'))]
for ax in g.axes.flatten():
ax.tick_params(labelbottom=True)
trxx=ax.get_title().split('|')[0].split('=')[1][1:-1]
mm=ax.get_title().split('|')[1].split('=')[1][1:]
data=AVG[(AVG['metric']==str(trxx))&(AVG['trx']==str(mm))]
annotator =Annotator(ax, data=data, x='time', y='value',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind',comparisons_correction='Bonferroni', text_format='simple', loc='inside')
annotator.apply_and_annotate()
# print(ax)
plt.tight_layout()
plt.show()
</code>
<code>
# B2graphs = load_list_of_dicts('data/gcn/B2X_Emore_ARG.pkl')
# # PS=pd.read_csv('data/gcn/comp_net/post_nest_clust_AA.txt',sep='\t',names=['G','C','AC','RAC','D','SB'])
# # LC=pd.DataFrame(columns=['Cl','LC','DC','BC','CC'])
G=B2graphs[12]
remove = [node for node,degree in dict(G.degree()).items() if degree < 3]
G.remove_nodes_from(remove)
top_nodes = {n for n, d in G.nodes(data=True) if d["bipartite"] == 0}
bottom_nodes = set(G) - top_nodes
tC=bipart(G,'clustering',top_nodes)
tLC=bipart(G,'latapy_clustering',top_nodes)
tNR=bipart(G,'node_redundancy',top_nodes)
tDC=bipart(G,'degree_centrality',top_nodes)
tBC=bipart(G,'betweenness_centrality',top_nodes)
tCC=bipart(G,'closeness_centrality',top_nodes)
# tMEC=bipart(G,'min_edge_cover',top_nodes)
tSBP=bipart(G,'spectral_bipartivity',top_nodes)
</code>
<code>
# LCC=LC.melt()
box_pairs=[]
for i in ['OTHER','LEVO','CLA']:
for j in ['_CT','_LC','_DC','_BC','_CC']:
LCC=LC.filter(like=i,axis=1)
LCC=LCC.filter(like=j,axis=1).melt()
LCC.variable=LCC.variable.str.split('_').str[1]+'_'+LCC.variable.str.split('_').str[2]+'_'+LCC.variable.str.split('_').str[3]
box_pairs.append(list(combinations(np.unique(LCC.variable), 2)))
box_pairs = list(chain(*box_pairs))
</code>
<code>
files=glob.glob('data/gcn/comp_net/*post_nest_clust_BIP2.txt')
for i,file in enumerate(files):
jj=os.path.basename(file).split('_post')[0]
if i==0:
LC=pd.read_csv(file,sep='\t',names=[jj+'_tC',jj+'_tLC',jj+'_tNR',jj+'_tDC',jj+'_tBC',jj+'_tCC',jj+'_tSBP',jj+'_bC',jj+'_bLC',jj+'_bNR',jj+'_bDC',jj+'_bBC',jj+'_bCC',jj+'_bSBP'])
else:
LC=pd.merge(LC,pd.read_csv(file,sep='\t',names=[jj+'_tC',jj+'_tLC',jj+'_tNR',jj+'_tDC',jj+'_tBC',jj+'_tCC',jj+'_tSBP',jj+'_bC',jj+'_bLC',jj+'_bNR',jj+'_bDC',jj+'_bBC',jj+'_bCC',jj+'_bSBP']),right_index=True,left_index=True,how='left')
# LC.columns=LC.columns.str.replace("_00", "_00ST")
# LC.columns=LC.columns.str.replace("_00STST", "_00ST")
LC.columns=LC.columns.str.replace("_03ST", "_02ST")
LCC=LC.melt()
LCC['trx']=LCC.variable.str.split('_').str[1]
LCC['time']=LCC.variable.str.split('_').str[2]
LCC['metric']=LCC.variable.str.split('_').str[3]
LCC=LCC[LCC.value!=0]
LCC=LCC.dropna()[~LCC.duplicated(keep='first')]
LCC.to_csv('data/gcn/comp_net/LCC_clust_BIP2.txt',sep='\t',header=True,index=True)
# LCC=LCC[(!LCC['metric'].contains'BSP')
# BSP, BC, NR
LCC=LCC[(~LCC.metric.str.contains('SBP'))&(~LCC.metric.str.contains('BC'))&(~LCC.metric.str.contains('NR'))]
ordered_days = sorted(LCC['trx'].unique())
g = sns.FacetGrid(LCC,col='trx',col_order=ordered_days,row='metric')
### change this to 4 ^
g.map(sns.boxplot,'time','value',palette='muted',order=['00ST','01ST','02ST'])
box_pairs=[(('00ST'),('01ST')),(('01ST'),('02ST')),(('00ST'),('02ST'))]
for ax in g.axes.flatten():
ax.tick_params(labelbottom=True)
trxx=ax.get_title().split('|')[0].split('=')[1][1:-1]
mm=ax.get_title().split('|')[1].split('=')[1][1:]
data=LCC[(LCC['metric']==str(trxx))&(LCC['trx']==str(mm))]
annotator =Annotator(ax, data=data, x='time', y='value',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind', text_format='simple', loc='inside',comparisons_correction='Bonferroni')
annotator.apply_and_annotate()
plt.tight_layout()
plt.show()
</code>
<code>
data=LCC[(LCC['trx']=='LEVO')&(LCC['metric']=='BC')]
stats.ttest_ind(data[data['time']=='00ST'].value,data[data['time']=='01ST'].value)
</code>
<code>
ddd=ccc[ccc['source'].str.contains('UniRef')]
ddd[['source','target']] = ddd[['target','source']]
ccc=ccc[~ccc['source'].str.contains('UniRef')].append(ddd)
ccc=ccc.sort_values(by=['source','target'])
</code>
<code>
AA=ccc.pivot('source',columns='target',values=ccc.columns[2]).fillna(0)#.round().astype('int').to_numpy())
# BB=ccc.pivot('source',columns='target',values=ccc.columns[2]).fillna(0).round().astype('int')#.to_numpy())M
</code>
<code>
tmp=pd.read_csv('tmp.txt',header=None)
tmp=tmp[0].str.split('.').str[1]
ddd=tmp.dropna()
len(ddd)
eee=pd.DataFrame(np.zeros((len(ddd),len(AA.columns))))
eee.index=ddd
eee.columns=AA.columns
AA=AA.append(eee)
AA=AA.reset_index().groupby('index').sum()
AA=AA.loc[:, (AA != 0).any(axis=0)]
AA=AA.loc[(AA != 0).any(axis=1),:]
M=((AA>0)*1).to_numpy()
</code>
## NestedNess
<code>
# M,*_ = NetworkGenerator.generate(500, 500, 4, bipartite=True, P=0.5, mu=0.5, alpha=2.5, min_block_size=0, fixedConn=False, link_density=2.45)
</code>
<code>
# M=np.corrcoef(np.transpose(M))
# M=((np.abs(M)>0.5)*1)
</code>
<code>
cols_degr=M.sum(axis=0)
row_degr=M.sum(axis=1)
R,C=M.shape #rows and cols
C_=extremal_bi.recursive_step(M,cols_degr,row_degr,.7,3,False)
Ci_=extremal_bi.recursive_step(M,cols_degr,row_degr,.7,3,True)
</code>
<code>
M[C_[0],:][:,C_[1]].shape
# A[0,:][:,0]
</code>
<code>
plt.imshow(M, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
N=M[np.argsort(np.sum(M,axis=1)),:][:,np.argsort(np.sum(M,axis=0))]
plt.imshow(N, cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
plt.imshow(M[C_[0],:][:,C_[1]], cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
plt.imshow(M[Ci_[0],:][:,Ci_[1]], cmap='hot', interpolation='nearest',aspect='auto')
plt.show()
</code>
<code>
aa=ccc.values
# nodes_cols = int(max(aa[j,1] for j in range(aa.shape[0]))+1)
# nodes_rows= int(max(aa[j,0] for j in range(aa.shape[0]))+1)
matrix=np.zeros((len(np.unique(ccc['source'])),len(np.unique(ccc['target']))),dtype='int')
</code>
<code>
t_12_CLA_00ST=pd.read_csv('nest/CLA_00ST/target_ARG035-00ST.txt',sep='\t',names=['C','protein'])
s_12_CLA_00ST=pd.read_csv('nest/CLA_00ST/source_ARG035-00ST.txt',sep='\t',names=['R','species'])
ib_12_CLA_00ST=pd.read_csv('nest/CLA_00ST/in-block_ARG035-00ST.txt',sep='\t',names=['R','C'])
mod_12_CLA_00ST=pd.read_csv('nest/CLA_00ST/modularity_ARG035-00ST.txt',sep='\t',names=['R','C'])
</code>
<code>
species=s_12_CLA_00ST['species'].dropna()
link_source=s_12_CLA_00ST['R'].dropna()
s_12_CLA_00ST=pd.DataFrame(species[link_source]).reset_index()
</code>
<code>
protein=t_12_CLA_00ST['protein'].dropna()
link_source=t_12_CLA_00ST['C'].dropna()
t_12_CLA_00ST=pd.DataFrame(protein[link_source]).reset_index()
</code>
<code>
[np.unique(ib_12_CLA_00ST['C']),
np.unique(ib_12_CLA_00ST['R'])]#.merge(t_12_CLA_00ST,left_on='C',right_on='index')
# IB#[~IB.duplicated(keep='first')]
</code>
<code>
ARG_meta=pd.read_excel('run/gcn/ARG_treatment_infor_modified.xlsx',index_col=0)
ARG_meta2=pd.read_excel('run/gcn/patients_Tx_batch3_for_DM.xlsx',index_col=None,skiprows=1,names=['id','group'])
relgene.columns=relgene.columns.str.replace("-00", "-00ST")
relgene.columns=relgene.columns.str.replace("-00STST", "-00ST")
relgene.columns=relgene.columns.str.split('-').str[0]+'-'+relgene.columns.str.split('-').str[1]
ARG_meta['id']=ARG_meta['id'].str.replace('-00ST','')
META=pd.concat([pd.DataFrame(ARG_meta[['id','group']]),ARG_meta2],ignore_index=True)
</code>
<code>
AA=np.loadtxt('nest/OTHER_00ST/32_OTHER_00ST.csv',dtype='int')
ccc=pd.DataFrame(AA)
ddd=ccc.sample(frac=0.2, replace=False, random_state=1)
rrr=ccc[~ccc.isin(ddd)].dropna(how='all')
ddd.reset_index(inplace=True)
del ddd['index']
sss=shuffle_net(ddd)
eee=pd.concat([rrr,sss])
aa=np.array(eee).astype(int)
</code>
<code>
# np.sum(eee.sort_values(['source','target']).reset_index()['ARG008-01ST-01-DNA_Abundance-RPKs']==ccc.sort_values(['source','target']).reset_index()['ARG008-01ST-01-DNA_Abundance-RPKs'])
</code>
<code>
#!python nestedness_analysis/structural_analysis.py nest/OTHER_01ST/ True True False False
</code>
<code>
# ccc.sort_values(['source','target'])==ddd.sort_values(['source','target'])
</code>
<code>
# C=pd.read_csv('~/data_structures_NQI_results.csv',header=0)#,'type','init'],sep='\t')
def create_data(path,rand):
C=pd.read_csv(path,header=0)#,'type','init'],sep='\t')
#### for randomized samples
C=C[C['name']!='name']
C['R']=C['R'].str.replace("False", "0")
# pd.unique(C['name'])
C=C[C['R']==rand]
del C['R']
# ####
C['type']=C['name'].str.split('_').str[1]+'_'+C['name'].str.split('_').str[2]
C['type']=C['type'].str.replace("_00", "_00ST")
# # C=C[~C['type'].str.contains("03")]
C['type']=C['type'].str.replace("_03ST", "_02ST")
C['type']=C['type'].str.replace("_00STST", "_00ST")
C['sample']=C['name'].str.split('_').str[0]
C=C[C['N']!='0.0']
# C=C[~C['name'].duplicated(keep='last')]
C=C[~C[['type','sample']].duplicated(keep='last')]
del C['name']
C.reset_index(inplace=True)
del C['index']
D=C.pivot(index='sample', columns='type', values=['N','I','Q'])
D=D.astype('float')
return D
def form_tests(data,var,level):
E0=data[var].reset_index()
E0=E0[['sample',level+'_00ST',level+'_01ST',level+'_02ST']]
E0['var']=var
return E0
def merge_form(data,level):
E0=form_tests(data,'N',level)
E1=form_tests(data,'I',level)
E2=form_tests(data,'Q',level)
E=E0.append(E1)
E=E.append(E2)
return E
def output_data(D):
E=merge_form(D,'CLA')
G=merge_form(D,'LEVO')
F=merge_form(D,'OTHER')
H=E.merge(G,on=['sample','var'])
H=H.merge(F,on=['sample','var'])
# H.set_index(['var','sample'],inplace=True)
# del H['var_x'],H['var_y']#,H0['type']
return H
# def prep_sub(H0):
# return H0
</code>
<code>
D0=create_data('randB_NQI_results.csv','0')
H0=output_data(D0)
# H0=prep_sub(H0)
D1=create_data('randB_NQI_results.csv','.2')
H1=output_data(D1)
# H1=prep_sub(H1)
D2=create_data('randB_NQI_results.csv','.99')
H2=output_data(D2)
# H2=prep_sub(H2)
</code>
<code>
H00=H0.melt(['sample','var'])
H11=H1.melt(['sample','var'])
H22=H2.melt(['sample','var'])
H01=H00.merge(H11,on=['sample','var','type'])
H01['diff']=H01['value_x']-H01['value_y']
H02=H00.merge(H22,on=['sample','var','type'])
H02['diff']=H02['value_x']-H02['value_y']
</code>
<code>
H12=H01.merge(H02,on=['sample','var','type'])
</code>
<code>
plt.figure(figsize=(6,6))
tmp=sns.scatterplot(data=H12,x='diff_x',y='diff_y',hue='var')#,bins=50,alpha=.3)
plt.xlim(-.2,.5)
plt.ylim(-.2,.5)
plt.plot([-.2, .5], [-.2, .5],ls='--',color='grey')
plt.xlabel('robust= Real - 20% shuffled')
plt.ylabel('random= Real - 99% shuffled')
</code>
<code>
FR0=pd.read_csv('SYNCSA_eval/rand0_FRall.csv')
FR0.rename(columns={'Unnamed: 0':'sample','cc.Simpson':'real_simpson','cc.FunRao':'real_Rao','cc.FunRedundancy':'real_FR','i':'type'},inplace=True)
FR0['sample']=FR0['sample'].str.split('_').str[0]
# FR_NIQ=FR.merge(NIQ,on=['sample','type'])
FR2=pd.read_csv('SYNCSA_eval/rand02_FRall.csv')
FR2.rename(columns={'Unnamed: 0':'sample','cc.Simpson':'robust_simpson','cc.FunRao':'robust_Rao','cc.FunRedundancy':'robust_FR','i':'type'},inplace=True)
FR2['sample']=FR2['sample'].str.split('_').str[0]
FR02=FR2.merge(FR0,on=['sample','type'])
FR9=pd.read_csv('SYNCSA_eval/rand9_FRall.csv')
FR9.rename(columns={'Unnamed: 0':'sample','cc.Simpson':'random_simpson','cc.FunRao':'random_Rao','cc.FunRedundancy':'random_FR','i':'type'},inplace=True)
FR9['sample']=FR9['sample'].str.split('_').str[0]
FR029=FR02.merge(FR9,on=['sample','type'])
</code>
<code>
# H12.columns=['sample','var','type','real','robust','tmp','tmp','random','tmp']
# del H12['tmp']
NIQ=H12.pivot(['sample','type'],columns='var')
NIQ.columns = [''.join(col) for col in NIQ.columns]
NIQ.reset_index(inplace=True)
# FR=pd.read_csv('SYNCSA_eval/rand0_FRall.csv')
# FR.rename(columns={'Unnamed: 0':'sample','cc.Simpson':'simpson','cc.FunRao':'Rao_Entropy','cc.FunRedundancy':'FR','i':'type'},inplace=True)
# FR['sample']=FR['sample'].str.split('_').str[0]
FR_NIQ=FR029.merge(NIQ,on=['sample','type'])
</code>
<code>
FR_NIQ.set_index(['sample','type'], inplace=True)
FR_NIQ=FR_NIQ.astype('float')
FR_NIQ.reset_index(inplace=True)
</code>
<code>
from sklearn import linear_model
from mpl_toolkits.mplot3d import Axes3D
FRNQ=FR_NIQ[['sample','type','real_FR','realQ','realN']].dropna(how='any')
######################################## Data preparation #########################################
# file = 'https://aegis4048.github.io/downloads/notebooks/sample_data/unconv_MV_v5.csv'
# df = pd.read_csv(file)
X = np.array(FRNQ[['realQ','realN']])#df[['Por', 'Brittle']].values.reshape(-1,2)
Y = np.array(FRNQ['real_FR'])#df['Prod']
######################## Prepare model data point for visualization ###############################
# x = np.array(FRNQ['FR'])
# y = np.array(FRNQ['realQ'])
# z = np.array(FRNQ['realN'])
x = X[:, 0]
y = X[:, 1]
z = Y
x_pred = np.linspace(0, 1, 10) # range of porosity values
y_pred = np.linspace(0, 1, 10) # range of brittleness values
# z_pred = np.linspace(0, 1, 10) # range of brittleness values
xx_pred, yy_pred = np.meshgrid(x_pred, y_pred)
model_viz = np.array([xx_pred.flatten(), yy_pred.flatten()]).T
################################################ Train #############################################
ols = linear_model.LinearRegression()
model = ols.fit(X, Y)
predicted = model.predict(model_viz)
############################################## Evaluate ############################################
r2 = model.score(X, Y)
############################################## Plot ################################################
plt.style.use('default')
fig = plt.figure(figsize=(12, 4))
ax1 = fig.add_subplot(131, projection='3d')
ax2 = fig.add_subplot(132, projection='3d')
ax3 = fig.add_subplot(133, projection='3d')
axes = [ax1, ax2, ax3]
for ax in axes:
ax.plot(x, y, z, color='k', zorder=15, linestyle='none', marker='.', alpha=0.5)
ax.scatter(xx_pred.flatten(), yy_pred.flatten(), predicted, facecolor=(0,0,0,0), s=20, edgecolor='#70b3f0')
ax.set_xlabel('nestedness (N)', fontsize=12)
ax.set_ylabel('modularity (Q)', fontsize=12)
ax.set_zlabel('FuncRed', fontsize=12)
ax.locator_params(nbins=4, axis='x')
ax.locator_params(nbins=5, axis='x')
# ax1.text2D(0.2, 0.32, 'aegis4048.github.io', fontsize=13, ha='center', va='center',
# transform=ax1.transAxes, color='grey', alpha=0.5)
# ax2.text2D(0.3, 0.42, 'aegis4048.github.io', fontsize=13, ha='center', va='center',
# transform=ax2.transAxes, color='grey', alpha=0.5)
# ax3.text2D(0.85, 0.85, 'aegis4048.github.io', fontsize=13, ha='center', va='center',
# transform=ax3.transAxes, color='grey', alpha=0.5)
ax1.view_init(elev=28, azim=120)
ax2.view_init(elev=4, azim=114)
ax3.view_init(elev=60, azim=165)
fig.suptitle('$R^2 = %.2f$' % r2, fontsize=20)
fig.tight_layout()
</code>
<code>
FR_II=FR_NIQ.filter(like='real',axis=1).melt('real_FR')
plt.figure(figsize=(6,6))
tmp=sns.scatterplot(data=FR_II,x='real_FR',y='value',hue='variable')
# plt.xlim(0,1)
# plt.ylim(0,1)
# plt.plot([0, 1], [0, 1],ls='--',color='grey')
</code>
<code>
import matplotlib.pyplot as plt
def annotate(data, **kws):
n = sum(FR_NIQ.filter(like='FR',axis=1)==FR_NIQ.filter(like='simpson',axis=1)-FR_NIQ.filter(like='Rao',axis=1))
ax = plt.gca()
ax.text(.1, .6, f"N = {n}", transform=ax.transAxes)
g = sns.FacetGrid(FR_NIQ.filter(like='FR',axis=1).melt(like='FR'), col="time")
g.map_dataframe(sns.scatterplot, x="total_bill", y="tip")
g.map_dataframe(annotate)
</code>
<code>
[sum(FR_NIQ['real_FR']==FR_NIQ['real_simpson']-FR_NIQ['real_Rao']),
sum(FR_NIQ['robust_FR']==FR_NIQ['robust_simpson']-FR_NIQ['robust_Rao']),
sum(FR_NIQ['random_FR']==FR_NIQ['random_simpson']-FR_NIQ['random_Rao'])]
</code>
<code>
def testprep(D):
E0=D['N'].reset_index()
E0=E0[['CLA_00ST','CLA_01ST','CLA_02ST']]
E0['type']='N'
E1=D['I'].reset_index()
E1=E1[['CLA_00ST','CLA_01ST','CLA_02ST']]
E1['type']='I'
E2=D['Q'].reset_index()
E2=E2[['CLA_00ST','CLA_01ST','CLA_02ST']]
E2['type']='Q'
E=E0.append(E1)
E=E.append(E2)
E0=D['N'].reset_index()
E0=E0[['LEVO_00ST','LEVO_01ST','LEVO_02ST']]
E0['type']='N'
E1=D['I'].reset_index()
E1=E1[['LEVO_00ST','LEVO_01ST','LEVO_02ST']]
E1['type']='I'
E2=D['Q'].reset_index()
E2=E2[['LEVO_00ST','LEVO_01ST','LEVO_02ST']]
E2['type']='Q'
F=E0.append(E1)
F=F.append(E2)
E0=D['N'].reset_index()
E0=E0[['OTHER_00ST','OTHER_01ST','OTHER_02ST']]
E0['type']='N'
E1=D['I'].reset_index()
E1=E1[['OTHER_00ST','OTHER_01ST','OTHER_02ST']]
E1['type']='I'
E2=D['Q'].reset_index()
E2=E2[['OTHER_00ST','OTHER_01ST','OTHER_02ST']]
E2['type']='Q'
G=E0.append(E1)
G=G.append(E2)
return E,F,G
</code>
<code>
from itertools import combinations
def ttest_run(c1, c2, E):
N=E[E['type']=='N'][[c1,c2]]#.dropna(how='any')
I=E[E['type']=='I'][[c1,c2]]#.dropna(how='any')
Q=E[E['type']=='Q'][[c1,c2]]#.dropna(how='any')
try:
resultsA = stats.ttest_rel(N[c1],N[c2],axis=0, nan_policy='omit', alternative='two-sided')
resultsB = stats.ttest_rel(I[c1],I[c2],axis=0, nan_policy='omit', alternative='two-sided')
resultsC = stats.ttest_rel(Q[c1],Q[c2],axis=0, nan_policy='omit', alternative='two-sided')
df = pd.DataFrame({'categ1': c1,
'categ2': c2,
'Ntstat': resultsA.statistic,
'Npvalue': resultsA.pvalue/2,
'Itstat': resultsB.statistic,
'Ipvalue': resultsB.pvalue/2,
'Qtstat': resultsC.statistic,
'Qpvalue': resultsC.pvalue/2,
'test':'paired'},
index = [0])
except:
resultsA = stats.ttest_rel(N[c1],N[c2])#, axis=0, nan_policy='omit', alternative='greater')
resultsB = stats.ttest_rel(I[c1],I[c2])#,axis=0, nan_policy='omit', alternative='greater')
resultsC = stats.ttest_rel(Q[c1],Q[c2])#,axis=0, nan_policy='omit', alternative='greater')
df = pd.DataFrame({'categ1': c1,
'categ2': c2,
'Ntstat': resultsA.statistic,
'Npvalue': resultsA.pvalue,
'Itstat': resultsB.statistic,
'Ipvalue': resultsB.pvalue,
'Qtstat': resultsC.statistic,
'Qpvalue': resultsC.pvalue,
'test':'ind'},
index = [0])
return df
df_list=pd.DataFrame()
for ii in [E,F,G]:
c = [ttest_run(i, j, ii) for i, j in combinations(ii.columns[:3].unique().tolist(), 2)]
df_list=df_list.append(c)
# final_df = pd.concat(df_list, ignore_index = True)
</code>
<code>
df_list=pd.DataFrame()
E,F,G=testprep(D0)
for ii in [E,F,G]:
c = [ttest_run(i, j, ii) for i, j in combinations(ii.columns[:3].unique().tolist(), 2)]
df_list=df_list.append(c)
df00=df_list
df00.set_index(['categ1','categ2'],inplace=True)#.astype('float')#-df02.astype('float')
del df00['test']
df00
</code>
<code>
df_list=pd.DataFrame()
E,F,G=testprep(D1)
for ii in [E,F,G]:
c = [ttest_run(i, j, ii) for i, j in combinations(ii.columns[:3].unique().tolist(), 2)]
df_list=df_list.append(c)
df02=df_list
df02.set_index(['categ1','categ2'],inplace=True)#.astype('float')#-df02.astype('float')
del df02['test']
df02
</code>
<code>
df_list=pd.DataFrame()
E,F,G=testprep(D2)
for ii in [E,F,G]:
c = [ttest_run(i, j, ii) for i, j in combinations(ii.columns[:3].unique().tolist(), 2)]
df_list=df_list.append(c)
df99=df_list
df99.set_index(['categ1','categ2'],inplace=True)#.astype('float')#-df02.astype('float')
del df99['test']
df99
</code>
<code>
cjj=(df00[['Npvalue','Ipvalue','Qpvalue']]-df02[['Npvalue','Ipvalue','Qpvalue']])
cjj.reset_index(inplace=True)
CJ=cjj.melt(['categ1','categ2'])
CJ.rename(columns={'value':'robust_val'},inplace=True)
cjj=(df00[['Npvalue','Ipvalue','Qpvalue']]-df99[['Npvalue','Ipvalue','Qpvalue']])
cjj.reset_index(inplace=True)
CJ2=cjj.melt(['categ1','categ2'])
CJ2.rename(columns={'value':'random_val'},inplace=True)
WWW=CJ.merge(CJ2,on=['categ1','categ2','variable'])
</code>
<code>
plt.figure(figsize=(6,6))
tmp=sns.histplot(data=WWW,x='robust_val',y='random_val',hue='variable',bins=30)
plt.xlim(-.2,.2)
plt.ylim(-.2,.2)
plt.xlabel('robustness= Real - 20% shuffled')
plt.ylabel('robustness= Real - 99% shuffled, ie random')
</code>
<code>
plt.figure(figsize=(6,6))
sns.scatterplot(data=WWW,x='robust_val',y='random_val',hue='variable')#,bins=30)
plt.xlim(-.2,.2)
plt.ylim(-.2,.2)
plt.plot([-.2, 1], [-.2, 1],ls='--',color='grey')
plt.xlabel('robustness= Real - 20% shuffled')
plt.ylabel('robustness= Real - 99% shuffled, ie random')
</code>
<code>
# del C['sample']
Cm=C.melt(['name','type'])
# del Cm['init']
Cm.value=pd.to_numeric(Cm.value)
Cm.sort_values(by=['type'],inplace=True)
</code>
<code>
box_pairs=[
# (('cc.FunRedundancy', 'CLA_00ST'), ('cc.FunRedundancy', 'LEVO_00ST')),
# (('cc.FunRedundancy', 'CLA_01ST'), ('cc.FunRedundancy', 'LEVO_01ST')),
# (('cc.FunRedundancy', 'CLA_02ST'), ('cc.FunRedundancy', 'LEVO_02ST')),
(('N', 'CLA_00ST'),( 'N', 'CLA_01ST')),
# (('N', 'CLA_00ST'), ('N', 'CLA_02ST')),
# (('N', 'CLA_01ST'), ('N', 'CLA_02ST')),
(('N', 'LEVO_00ST'),( 'N', 'LEVO_01ST')),
# (('N', 'LEVO_00ST'), ('N', 'LEVO_02ST')),
# (('N', 'LEVO_01ST'), ('N', 'LEVO_02ST')),
# (('N', 'CLA_00ST'), ('N', 'LEVO_01ST')),
# (('N', 'CLA_01ST'), ('N', 'OTHER_02ST')),
# (('N', 'OTHER_00ST'), ('N', 'LEVO_02ST')),
(('I', 'CLA_00ST'),( 'I', 'CLA_01ST')),
# (('I', 'CLA_00ST'), ('I', 'CLA_02ST')),
# (('I', 'CLA_01ST'), ('I', 'CLA_02ST')),
(('I', 'LEVO_00ST'),( 'I', 'LEVO_01ST')),
# (('I', 'LEVO_00ST'), ('I', 'LEVO_02ST')),
# (('I', 'LEVO_01ST'), ('I', 'LEVO_02ST')),
# (('I', 'CLA_00ST'), ('I', 'LEVO_01ST')),
# (('I', 'CLA_01ST'), ('I', 'OTHER_02ST')),
# (('I', 'OTHER_00ST'), ('I', 'LEVO_02ST')),
(('Q', 'CLA_00ST'),( 'Q', 'CLA_01ST')),
# (('Q', 'CLA_00ST'), ('Q', 'CLA_02ST')),
# (('Q', 'CLA_01ST'), ('Q', 'CLA_02ST')),
(('Q', 'LEVO_00ST'),( 'Q', 'LEVO_01ST')),
# (('Q', 'LEVO_00ST'), ('Q', 'LEVO_02ST')),
# (('Q', 'LEVO_01ST'), ('Q', 'LEVO_02ST')),
# (('Q', 'CLA_00ST'), ('Q', 'LEVO_01ST')),
# (('Q', 'CLA_01ST'), ('Q', 'OTHER_02ST')),
# (('Q', 'OTHER_00ST'), ('Q', 'LEVO_02ST'))
]
# (('cc.FunRedundancy', 'OTHER_00ST'),( 'cc.FunRedundancy', 'OTHER_01ST'))]#,
# (('cc.FunRedundancy', 'CLA_00ST'), ('cc.FunRedundancy', 'CLA_02ST')),]#,
plt.figure(figsize=(8,4))
# B.dropna(inplace=True)
# B=B.reset_index()
# C=B[B[typ]>0]
ax=sns.boxplot(data=www,y='value',x='variable',hue='type')
# add_stat_annotation(ax, data=Cm, x='variable', y='value',hue='type',
# box_pairs=box_pairs,#comparisons,
# test='t-test_paired', text_format='star', loc='outside', verbose=0)
# annotator =Annotator(ax, data=Cm, x='variable', y='value',hue='type',pairs=box_pairs)#comparisons,
# annotator.configure(test='Levene', text_format='star', loc='outside')
# annotator.apply_and_annotate()
# t-test_ind, t-test_welch, t-test_paired, Mann-Whitney, Mann-Whitney-gt, Mann-Whitney-ls, Levene, Wilcoxon, Kruskal
###change to paired when all samples are finished
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
# plt.savefig(".png",dpi=300,bbox_inches = "tight")
</code>
<code>
ax=sns.scatterplot(data=C,y='N',x='Q',hue='I')
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
</code>
# Redundancy = taxon diversity - functional diversity
<code>
relgene=pd.read_csv('all_arg_subset_genefamilies-cpm.tsv',sep='\t',nrows=1)
graphs = load_list_of_dicts('data/gcn/NX_Emore_ARG.pkl')
ARG_meta=pd.read_excel('run/gcn/ARG_treatment_infor_modified.xlsx',index_col=0)
ARG_meta2=pd.read_excel('run/gcn/patients_Tx_batch3_for_DM.xlsx',index_col=None,skiprows=1,names=['id','group'])
relgene.columns=relgene.columns.str.replace("-00", "-00ST")
relgene.columns=relgene.columns.str.replace("-00STST", "-00ST")
relgene.columns=relgene.columns.str.split('-').str[0]+'-'+relgene.columns.str.split('-').str[1]
ARG_meta['id']=ARG_meta['id'].str.replace('-00ST','')
META=pd.concat([pd.DataFrame(ARG_meta[['id','group']]),ARG_meta2],ignore_index=True)
</code>
<code>
JEFF=pd.DataFrame()
deg_rand=0
# def makeSYNCSAnet(graphs,JEFF,META,deg_rand):
# for i,net in tqdm.tqdm(enumerate(BX_graphs)):
# for ii,i in tqdm():
for ii,i in tqdm(enumerate(relgene.columns[1:])):
ccc=nx.convert_matrix.to_pandas_edgelist(graphs[ii])
ee=nx.convert_matrix.to_pandas_edgelist(graphs[ii])
# cc['weight']=np.random.randn(len(cc))
pww=i
j=(i.split('-')[1])
i=(i.split('-')[0])
try:
rrr=str(META[META['id']==i].index.item())+'_'+str(META[META['id']==i]['group'].item())+'_'+str(j)
ccc.rename(columns={ccc.columns[2]:rrr},inplace=True)
ddd=ccc[ccc['source'].str.contains('UniRef')]
ddd[['source','target']] = ddd[['target','source']]
ccc=ccc[~ccc['source'].str.contains('UniRef')].append(ddd)
if deg_rand!=0:
aa=pd.DataFrame(ccc)
pcc=aa.sample(frac=np.float(deg_rand), replace=False, random_state=1) ##degree randomized
pol=aa[~aa.isin(pcc)].dropna(how='all')
pcc.reset_index(inplace=True)
del pcc['index']
lll=shuffle_net(pcc)
ccc=pd.concat([pol,lll])
del aa,pol,pcc,lll
# a,b=pd.factorize(ccc['source'])
# c,d=pd.factorize(ccc['target'])
# rrr=pd.DataFrame()
# rrr['from']=a
# rrr['to']=c
# rrr['value']=1
# sss=str(META[META['id']==i]['group'].item())+'_'+str(j)
# Path('~/nest/'+sss).mkdir(parents=True, exist_ok=True)
# rrr[['from','to','value']].to_csv('~/nest/'+sss+'/'+str(ccc.columns[2])+'.csv',sep=' ',index=False,header=False)
# ee.rename(columns={ee.columns[2]:sss},inplace=True)
# print(ii)
if ii==0:
dd=ccc
# ff=ee
else:
dd=dd.merge(ccc,on=['source','target'],how='outer')
# ff=ff.merge(ee,on=['source','target'],how='outer')
# return dd/
del ddd,rrr,ccc,ee
except:
print('no match for '+str(i))
</code>
<code>
names=pd.unique(dd.columns.str.split('_').str[1]+'_'+dd.columns.str.split('_').str[2])[1:]
for i in tqdm(names):
# def group4SYNCSA(i,dd,DR):
# ff.columns = ff.columns.str.strip('_x')
# ff.columns = ff.columns.str.strip('_y')
# i=i.split('_')[1]+'_'+i.split('_')[2]
ff=dd.loc[:,dd.columns.str.contains(i)]
ff[['source','target']]=dd[['source','target']]
ff=ff[ff['source'].str.contains('s__')]
ff=ff[ff['target'].str.contains('UniRef')]
comm=ff.groupby('source').sum().transpose()
comm.to_csv('~/SYNCSA_eval/'+str(deg_rand)+'_rand_comm_'+i+'.csv')
ff.reset_index(inplace=True)
ff.set_index(['source', 'target'], inplace=True)
del ff['index']
ff.columns=(ff.columns.str.split('_').str[1]+'_'+ff.columns.str.split('_').str[2])
gg=ff.groupby(by=ff.columns, axis=1).sum()
# traits=gg[[i]].reset_index().pivot('source','target',i).dropna(how='all',axis=1).replace(np.nan,0)
traits=gg[[i]].reset_index().groupby(['source','target']).mean().reset_index().pivot('source','target',i).dropna(how='all',axis=1).replace(np.nan,0)
traits.to_csv('~/SYNCSA_eval/'+str(deg_rand)+'_rand_trait_'+i+'.csv')
</code>
### process in R
<code>
!Rscript ~/run/gcn/calc_FR.r -d 0
!Rscript ~/run/gcn/calc_FR.r -d 0.2
!Rscript ~/run/gcn/calc_FR.r 0.99
# library('SYNCSA')
# files <- data.frame('CLA_00ST','CLA_01ST','CLA_02ST','LEVO_00ST','LEVO_01ST',
# 'LEVO_02ST','OTHER_00ST','OTHER_01ST','OTHER_02ST')
# for (i in files){
# trait=read.csv(paste('~/SYNCSA_eval/trait_',i,'.csv',sep=''))
# comm=read.csv(paste('~/SYNCSA_eval/comm_',i,'.csv',sep=''))
# row.names(trait)<-trait$source
# trait$source<-NULL
# row.names(comm)<-comm$X
# comm$X<-NULL
# cc=rao.diversity(comm, traits =trait)
# write.csv(data.frame(cc$Simpson,cc$FunRao,cc$FunRedundancy,i),paste('~/SYNCSA_eval/FR_',i,'.csv',sep=''))
# # }
</code>
<code>
!cat SYNCSA_eval/0rand_FR* >SYNCSA_eval/rand00_FRall.csv
!cat SYNCSA_eval/0.2rand_FR* >SYNCSA_eval/rand02_FRall.csv
!cat SYNCSA_eval/0.9rand_FR* >SYNCSA_eval/rand9_FRall.csv
</code>
<code>
FR=pd.read_csv('SYNCSA_eval/rand0_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
del FR['i']#,FR['Unnamed: 0'],
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
FRm00=FR.melt('index')
FRm00.value=pd.to_numeric(FRm00.value)
FR=pd.read_csv('SYNCSA_eval/rand02_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
del FR['i']#,FR['Unnamed: 0'],
FRm02=FR.melt('index')
FRm02.value=pd.to_numeric(FRm02.value)
FR=pd.read_csv('SYNCSA_eval/rand9_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
del FR['i']#,FR['Unnamed: 0'],
FRm9=FR.melt('index')
FRm9.value=pd.to_numeric(FRm9.value)
</code>
<code>
box_pairs=[
(('cc.Simpson', 'CLA_00ST'),( 'cc.Simpson', 'CLA_01ST')),
(('cc.Simpson', 'CLA_00ST'), ('cc.Simpson', 'CLA_02ST')),
(('cc.Simpson', 'CLA_01ST'), ('cc.Simpson', 'CLA_02ST')),
(('cc.Simpson', 'LEVO_00ST'),( 'cc.Simpson', 'LEVO_01ST')),
(('cc.Simpson', 'LEVO_00ST'), ('cc.Simpson', 'LEVO_02ST')),
(('cc.Simpson', 'LEVO_01ST'), ('cc.Simpson', 'LEVO_02ST')),
(('cc.Simpson', 'OTHER_00ST'),( 'cc.Simpson', 'OTHER_01ST')),
(('cc.Simpson', 'OTHER_00ST'), ('cc.Simpson', 'OTHER_02ST')),
(('cc.Simpson', 'OTHER_01ST'), ('cc.Simpson', 'OTHER_02ST')),
(('cc.FunRedundancy', 'CLA_00ST'),( 'cc.FunRedundancy', 'CLA_01ST')),
(('cc.FunRedundancy', 'CLA_00ST'), ('cc.FunRedundancy', 'CLA_02ST')),
(('cc.FunRedundancy', 'CLA_01ST'), ('cc.FunRedundancy', 'CLA_02ST')),
(('cc.FunRedundancy', 'LEVO_00ST'),( 'cc.FunRedundancy', 'LEVO_01ST')),
(('cc.FunRedundancy', 'LEVO_00ST'), ('cc.FunRedundancy', 'LEVO_02ST')),
(('cc.FunRedundancy', 'LEVO_01ST'), ('cc.FunRedundancy', 'LEVO_02ST')),
(('cc.FunRedundancy', 'OTHER_00ST'),( 'cc.FunRedundancy', 'OTHER_01ST')),
(('cc.FunRedundancy', 'OTHER_00ST'), ('cc.FunRedundancy', 'OTHER_02ST')),
(('cc.FunRedundancy', 'OTHER_01ST'), ('cc.FunRedundancy', 'OTHER_02ST')),
(('cc.FunRao', 'CLA_00ST'),( 'cc.FunRao', 'CLA_01ST')),
(('cc.FunRao', 'CLA_00ST'), ('cc.FunRao', 'CLA_02ST')),
(('cc.FunRao', 'CLA_01ST'), ('cc.FunRao', 'CLA_02ST')),
(('cc.FunRao', 'LEVO_00ST'),( 'cc.FunRao', 'LEVO_01ST')),
(('cc.FunRao', 'LEVO_00ST'), ('cc.FunRao', 'LEVO_02ST')),
(('cc.FunRao', 'LEVO_01ST'), ('cc.FunRao', 'LEVO_02ST')),
(('cc.FunRao', 'OTHER_00ST'),( 'cc.FunRao', 'OTHER_01ST')),
(('cc.FunRao', 'OTHER_00ST'), ('cc.FunRao', 'OTHER_02ST')),
(('cc.FunRao', 'OTHER_01ST'), ('cc.FunRao', 'OTHER_02ST'))
]
# (('cc.FunRedundancy', 'OTHER_00ST'),( 'cc.FunRedundancy', 'OTHER_01ST'))]#,
# (('cc.FunRedundancy', 'CLA_00ST'), ('cc.FunRedundancy', 'CLA_02ST')),]#,
</code>
<code>
# def plt_plect(B,typ):
plt.figure(figsize=(8,4))
# B.dropna(inplace=True)
# B=B.reset_index()
# C=B[B[typ]>0]
ax=sns.boxplot(data=FRm00,y='value',x='variable',hue='index')
annotator =Annotator(ax, data=FRm00, x='variable', y='value',hue='index',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind', text_format='star', loc='outside')
annotator.apply_and_annotate()
# add_stat_annotation(ax, data=FRm, x='variable', y='value',hue='index',
# box_pairs=box_pairs,#comparisons,
# test='t-test_ind', text_format='star', loc='outside', verbose=0)
###change to paired when all samples are finished
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
plt.savefig("FR00.png",dpi=300,bbox_inches = "tight")
</code>
<code>
FRm00['trx']=FRm00['index'].str.split('_').str[0]
FRm00['time']=FRm00['index'].str.split('_').str[1]
FRm00['metric']=FRm00['variable'].str.split('.').str[1]
</code>
<code>
FRm00
</code>
<code>
# LCC=LC.melt()
# LCC['trx']=LCC.variable.str.split('_').str[1]
# LCC['time']=LCC.variable.str.split('_').str[2]
# LCC['metric']=LCC.variable.str.split('_').str[3]
# LCC=LCC[LCC.value!=0]
ordered_days = sorted(FRm00['trx'].unique())
g = sns.FacetGrid(FRm00,col='trx',col_order=ordered_days,row='metric')
# change this to 4 ^
g.map(sns.boxplot,'time','value',palette='muted')
box_pairs=[(('00ST'),('01ST')),(('01ST'),('02ST')),(('00ST'),('02ST'))]
for ax in g.axes.flatten():
ax.tick_params(labelbottom=True)
trxx=ax.get_title().split('|')[0].split('=')[1][1:-1]
mm=ax.get_title().split('|')[1].split('=')[1][1:]
data=FRm00[(FRm00['metric']==str(trxx))&(FRm00['trx']==str(mm))]
annotator =Annotator(ax, data=data, x='time', y='value',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind',comparisons_correction='Bonferroni', text_format='simple', loc='inside')
annotator.apply_and_annotate()
# print(ax)
plt.tight_layout()
plt.show()
</code>
<code>
trxx=ax.get_title().split('|')[0].split('=')[1][1:-1]
mm=ax.get_title().split('|')[1].split('=')[1][1:]
data=FRm00[(FRm00['metric']==str(trxx))&(FRm00['trx']==str(mm))]
</code>
<code>
# def plt_plect(B,typ):
plt.figure(figsize=(8,4))
# B.dropna(inplace=True)
# B=B.reset_index()
# C=B[B[typ]>0]
ax=sns.boxplot(data=FRm02,y='value',x='variable',hue='index')
annotator =Annotator(ax, data=FRm02, x='variable', y='value',hue='index',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind', text_format='star', loc='outside')
annotator.apply_and_annotate()
# add_stat_annotation(ax, data=FRm, x='variable', y='value',hue='index',
# box_pairs=box_pairs,#comparisons,
# test='t-test_ind', text_format='star', loc='outside', verbose=0)
###change to paired when all samples are finished
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
plt.savefig("FR02.png",dpi=300,bbox_inches = "tight")
</code>
<code>
# def plt_plect(B,typ):
plt.figure(figsize=(8,4))
# B.dropna(inplace=True)
# B=B.reset_index()
# C=B[B[typ]>0]
ax=sns.boxplot(data=FRm9,y='value',x='variable',hue='index')
annotator =Annotator(ax, data=FRm9, x='variable', y='value',hue='index',pairs=box_pairs,verbose=0)#comparisons,
annotator.configure(test='t-test_ind', text_format='star', loc='outside')
annotator.apply_and_annotate()
# add_stat_annotation(ax, data=FRm, x='variable', y='value',hue='index',
# box_pairs=box_pairs,#comparisons,
# test='t-test_ind', text_format='star', loc='outside', verbose=0)
###change to paired when all samples are finished
plt.legend(loc='upper left', bbox_to_anchor=(1.03, 1))
plt.savefig("FR9.png",dpi=300,bbox_inches = "tight")
</code>
<code>
# # cat rand.2_FR* >rand02_FRall.csv
FR=pd.read_csv('SYNCSA_eval/rand0_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
# FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
del FR['i']#,FR['Unnamed: 0'],
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
FRm00=FR.melt('index')
FRm00.value=pd.to_numeric(FRm00.value)
# cat .2rand_FR* >rand02_FRall.csv
FR=pd.read_csv('SYNCSA_eval/rand02_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
# FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
del FR['i']#,FR['Unnamed: 0'],
FRm02=FR.melt('index')
FRm02.value=pd.to_numeric(FRm02.value)
# # cat rand.2_FR* >rand02_FRall.csv
FR=pd.read_csv('SYNCSA_eval/rand9_FRall.csv')
FR=FR[FR['Unnamed: 0']!='NaN']
FR=FR[FR['Unnamed: 0']!=np.nan]
FR=FR[FR['cc.Simpson']!='cc.Simpson']
# FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
FR.rename(columns={'Unnamed: 0':'index'},inplace=True)
del FR['i']#,FR['Unnamed: 0'],
FRm9=FR.melt('index')
FRm9.value=pd.to_numeric(FRm9.value)
</code>
<code>
FR02=FRm00.merge(FRm02,on=['index','variable'])
FR02['diff']=FR02['value_x']-FR02['value_y']
FR09=FRm00.merge(FRm9,on=['index','variable'])
FR09['diff']=FR09['value_x']-FR09['value_y']
FR29=FR02[['index','variable','diff']].merge(FR09[['index','variable','diff']],on=['index','variable'])
</code>
<code>
plt.figure(figsize=(6,6))
tmp=sns.scatterplot(data=FR29,x=(FR29['diff_x']),y=(FR29['diff_y']),hue='variable')#,bins=50,alpha=.3)
plt.xlim(-.2,1)
plt.ylim(-.2,1)
plt.plot([-.2, 1], [-.2, 1],ls='--',color='grey')
plt.xlabel('robustness= Real - 20% shuffled')
plt.ylabel('random= Real - 90% shuffled')
</code>
# ttest diff Ab
<code>
names=('CLA_00ST','CLA_01ST','CLA_02ST','LEVO_00ST','LEVO_01ST',
'LEVO_02ST','OTHER_00ST','OTHER_01ST','OTHER_02ST')
FR=pd.DataFrame()
for ii,i in enumerate(names):
j=pd.read_csv('SYNCSA_eval/0_rand_comm_'+i+'.csv')
if ii==0:
FR=(j)
else:
FR=FR.append(j)
FR['Unnamed: 0']=FR['Unnamed: 0'].str.split('_').str[1]+'_'+FR['Unnamed: 0'].str.split('_').str[2]
FR_00Ab=FR.melt('Unnamed: 0')
FR_00Ab=FR_00Ab[FR_00Ab['value']!=0]
</code>
<code>
FR_00Ab['spec']=FR_00Ab['variable'].str.split('_').str[2]
# spFR_00Ab=FR_00Ab.groupby(['spec','Unnamed: 0']).sum()
# spFR_00Ab.reset_index(inplace=True)
FR_00Ab['trx']=FR_00Ab['Unnamed: 0'].str.split('_').str[0]
FR_00Ab['time']=FR_00Ab['Unnamed: 0'].str.split('_').str[1].str.split('ST').str[0]
</code>
<code>
FR_00Ab[FR_00Ab['spec'].isin(np.unique(FR_00Ab['spec'])[0:10])]
</code>
<code>
rr=pd.DataFrame(columns=['spec','p01','p02','p12'])
# rr=[]
# cc=pd.DataFrame(columns=['spec','p01','p02','p12'])
for j in np.unique(FR_00Ab['trx']):
for i in np.unique(FR_00Ab['spec']):
data=FR_00Ab[FR_00Ab['spec']==i]
data=data[data['trx']==j]
bb=stats.ttest_ind(data[data['time']=='00']['value'],data[data['time']=='01']['value'],axis=0, nan_policy='omit', alternative='two-sided')
dd=stats.ttest_ind(data[data['time']=='00']['value'],data[data['time']=='02']['value'],axis=0, nan_policy='omit', alternative='two-sided')
ee=stats.ttest_ind(data[data['time']=='01']['value'],data[data['time']=='02']['value'],axis=0, nan_policy='omit', alternative='two-sided')
cc = pd.DataFrame({'spec': i,
'p01': bb.pvalue,
'p02': dd.pvalue,
'p12': ee.pvalue,
'type':j},
index = [0])
rr=rr.append(cc)#,ignore_index=True)
</code>
<code>
rrr=rr[~((rr['spec']=='Streptococcus')&(rr['type']=='OTHER'))]
rrr=rrr.melt(['spec','type'])
rrr=rrr[(rrr['value']<.05)].sort_values('type')
rrr.pivot(['spec','type'],columns='variable').dropna(how='all')
</code>
<code>
rrr.groupby(['spec','type']).count()
</code>
<code>
FR_00Ab['value']=np.log(FR_00Ab['value'])
</code>
<code>
FRcalc=FR_00Ab.groupby(['spec','trx','time']).sum().reset_index()
FRcalc=FRcalc.pivot(['spec','trx'],columns='time').reset_index()
# rrr.pivot(['spec','type'],columns='variable').dropna(how='all')
FRcalc.columns=['spec','trx','00','01','02']
</code>
<code>
AA=FRcalc[FRcalc['trx']=='CLA']
</code>
<code>
CLA_hill=FRcalc[(FRcalc['00']<FRcalc['01']/2)&(FRcalc['01']/2>FRcalc['02'])]
CLA_hill=CLA_hill.melt(['spec','trx'])
# sns.lineplot(data=CLA_hill, x="variable", y="value", hue="spec")
sns.relplot(
data=CLA_hill, x="variable", y="value",
col="trx", hue="spec",#, style="event",
kind="line",col_order=['CLA','LEVO','OTHER']#,x_order=['00ST','01ST','02ST']
)
</code>
<code>
CLA_val=FRcalc[(FRcalc['00']>FRcalc['01'])&(FRcalc['01']<FRcalc['02'])]
CLA_val=CLA_val.melt(['spec','trx'])
# sns.lineplot(data=CLA_hill, x="variable", y="value", hue="spec")
sns.relplot(
data=CLA_val, x="variable", y="value",
col="trx", hue="spec",#, style="event",
kind="line",col_order=['CLA','LEVO','OTHER']#,x_order=['00ST','01ST','02ST']
)
</code>
<code>
plt.figure(figsize=(10,8))
# sns.lineplot(data=FR_00Ab, x="time", y="value", hue="trx", style="spec")
# FR_00Ab=FR_00Ab[FR_00Ab['trx']=='CLA']
for i in range(1,117,10):
sns.relplot(
data=FR_00Ab[FR_00Ab['spec'].isin(np.unique(FR_00Ab['spec'])[i:i+10])], x="time", y=(FR_00Ab["value"]),
col="trx", hue="spec",#, style="event",
kind="line",col_order=['CLA','LEVO','OTHER']#,x_order=['00ST','01ST','02ST']
)
# plt.savefig("data/gcn/img/spec_trx_012"+str(i)+".png",dpi=300,bbox_inches = "tight")
</code>
<code>
spFR_00Ab.reset_index(inplace=True)
spFR_0=spFR_00Ab[spFR_00Ab['Unnamed: 0'].str.contains('00ST')]
spFR_1=spFR_00Ab[spFR_00Ab['Unnamed: 0'].str.contains('01ST')]
spFR_2=spFR_00Ab[spFR_00Ab['Unnamed: 0'].str.contains('02ST')]
spFR_01=spFR_0.merge(spFR_1,on=['spec','trx'])
spFR_02=spFR_0.merge(spFR_2,on=['spec','trx'])
spFR_12=spFR_1.merge(spFR_2,on=['spec','trx'])
</code>
<code>
plt.figure(figsize=(8,8))
sns.scatterplot(data=spFR_01, x="value_x", y="value_y", hue="trx")
plt.xlim(-1,30000)
plt.ylim(-1,30000)
plt.xlabel('00ST - total spec Abs')
plt.ylabel('01ST - total spec Abs')
</code>
# Arg Ab nestedness
<code>
names=('CLA_00ST','CLA_01ST','CLA_02ST','LEVO_00ST','LEVO_01ST',
'LEVO_02ST','OTHER_00ST','OTHER_01ST','OTHER_02ST')
rr=pd.DataFrame(columns=['N','Q','I','name'])
for ii,i in enumerate(names):
FR=pd.read_csv('SYNCSA_eval/0_rand_comm_'+i+'.csv')
FR.rename(columns={'Unnamed: 0':'indv'},inplace=True)
aa=FR.dropna().melt('indv')
labels1,levels1=pd.factorize(aa['indv'])
aa['indv']=labels1
labels2,levels2=pd.factorize(aa['variable'])
aa['variable']=labels2
# aa['value']=1
aa=aa[aa['value']>0]
aa['value']=1
aa=np.array(aa)
# FR0.to_csv('nest/FR_test/'+i+'.txt',sep='\t',header=False,index=False)
nodes_cols = int(max(aa[j,1] for j in range(aa.shape[0]))+1)
nodes_rows= int(max(aa[j,0] for j in range(aa.shape[0]))+1)
M=np.zeros((nodes_rows,nodes_cols),dtype='int')
for j in range(aa.shape[0]):
M[aa[j,0],aa[j,1]] = 1
cols_degr=M.sum(axis=0)
row_degr=M.sum(axis=1)
R,C=M.shape #rows and cols
#Nestednes
# In-block nestedness with B=1
Cn_=[np.repeat(1, R),np.repeat(1, C)]
max_blockN=max(max(Cn_[0]),max(Cn_[1]))+1
lambdasN=extremal_bi.call_lambda_i(M,cols_degr,row_degr,Cn_[1],Cn_[0],max_blockN,True)
nestedness_=extremal_bi.calculate_Fitness(M,cols_degr,row_degr,lambdasN[0],lambdasN[1],True)
#Modularity Extremal
C_=extremal_bi.recursive_step(M,cols_degr,row_degr,.7,3,False)
max_blockQ=max(max(C_[0]),max(C_[1]))+1
lambdasQ=extremal_bi.call_lambda_i(M,cols_degr,row_degr,C_[1],C_[0],max_blockQ,False)
Q_=extremal_bi.calculate_Fitness(M,cols_degr,row_degr,lambdasQ[0],lambdasQ[1],False)
# Inblock nestedness extremal
Ci_=extremal_bi.recursive_step(M,cols_degr,row_degr,.7,3,True)
max_blockI=max(max(Ci_[0]),max(Ci_[1]))+1
lambdasI=extremal_bi.call_lambda_i(M,cols_degr,row_degr,Ci_[1],Ci_[0],max_blockI,True)
I_=extremal_bi.calculate_Fitness(M,cols_degr,row_degr,lambdasI[0],lambdasI[1],True)
[nestedness_,Q_,I_,ii]
cc = pd.DataFrame({'N': nestedness_,
'Q': Q_,
'I': I_,
'name': i},
index = [0])
rr=rr.append(cc)#,ignore_index=True)
</code>
<code>
rr
</code>
<code>
# rr['spec']=rr['variable'].str.split('_').str[2]
# spFR_00Ab=FR_00Ab.groupby(['spec','Unnamed: 0']).sum()
# spFR_00Ab.reset_index(inplace=True)
rr['trx']=rr['name'].str.split('_').str[0]
rr['time']=rr['name'].str.split('_').str[1].str.split('ST').str[0]
del rr['name']
rr00=rr.melt(['time','trx'])
</code>
<code>
plt.figure(figsize=(10,8))
# sns.lineplot(data=FR_00Ab, x="time", y="value", hue="trx", style="spec")
# for i in range(1,117,10):
sns.relplot(
data=rr00, x="time", y='value',
col="variable", hue="trx",#, style="event",
kind="line",hue_order=['CLA','LEVO','OTHER']#,x_order=['00ST','01ST','02ST']
)
# plt.savefig("data/gcn/img/spec_trx_012"+str(i)+".png",dpi=300,bbox_inches = "tight")
</code>
<code>
!python nestedness_analysis/structural_analysis.py nest/FR_test/ True True 0
</code>
<code>
sys.path.insert(1, './run/gcn/')
import gcn_func
importlib.reload(sys.modules['gcn_func'])
from gcn_func import research_orthologs
norm_Px=pd.read_csv('data/Pipeline_consolidate_220301/Norm_Px.txt',sep='\t')
PS=pd.DataFrame(columns=['mus','homo','spec'])
for i in norm_Px['Protein']:
try:
P,S=research_orthologs(str(i),'Homo sapiens')
except:
print(i)
cc = pd.DataFrame({'mus': i,
'homo': P,
'spec': S},
index = [0])
PS=PS.append(cc)
cc.to_csv('mus_homo_int2.txt',sep='\t',mode='a')
PS.to_csv('mus_homo2.txt',sep='\t')
</code>
|
{
"filename": "gcn_1.ipynb",
"repository": "dcolinmorgan/gcn",
"query": "transformed_from_existing",
"size": 303642,
"sha": ""
}
|
# lab_symintro_1.ipynb
Repository: nicollsf/eee2047s-notebooks
# Symbolic math introduction
Symbolic mathematics is a maturing technology that lets a computer do maths using symbolic manipulation rather than numerical computation. Python has support for symbolic computation via the "sympy" package.
The sympy documentation can be found at http://docs.sympy.org/latest/index.html. The PDF version is just under 2000 pages long, which is quite frightening.
Nonetheless, this notebook will introduce some of the basics of symbolic math using Python. In particular we will see how to define functions of symbolic variables, and differentiate and integrate them symbolically.
Some other good examples of sympy in use are at http://www.cfm.brown.edu/people/dobrush/am33/SymPy/index.html and https://github.com/sympy/sympy/wiki/Quick-examples. The first one in particular deals with differential equations, which will be used in subsequent computer assignments.
## Basic differentiation
The cell below imports the symbolic math package, and defines two symbolic variables `x` and `y`. A symbolic function $f(x,y) = (x^2-2x+3)/y$ is then defined and printed.
<code>
import sympy as sp #import the sympy library under the name 'sp'
#?sp.integrate()
x, y = sp.symbols('x y');
f = (x**2 - 2*x + 3)/y;
print(f);
</code>
<code>
# protip: you can make your outputs look sexy with LaTex:
from IPython.display import display
sp.init_printing() # initializes pretty printing. Only needs to be run once.
display(f)
</code>
Note that `f` here is a symbol representing a function. It would be nice if the notation made it explicit that it's actually a function of $x$ and $y$, namely `f(x,y)`, but that's not how it works. However, we can query the free variables:
<code>
f.free_symbols
</code>
We can get sympy to find a symbolic expression for the partial derivative of $f(x,y)$ with respect to $y$:
<code>
fpy = sp.diff(f, y)
fpy
</code>
To evaluate this derivative at some particular values $x=\pi$ and $y=2$ we can substitute into the symbolic expression:
<code>
fpyv = fpy.subs([(x, sp.pi), (y, 2)])
fpyv
</code>
Notice though that this is still a symbolic expression. It can be evaluated using the "evalf" method, which finally returns a number:
<code>
fpyv.evalf()
</code>
## More advanced differentiation
Symbolic expressions can be manipulated. For example we can define $g(t) = f(x(t), y(t))$, which in this case given above means
$$g(t) = (x(t)^2-2x(t)+3)/y(t),$$
and find its derivative with respect to time.
<code>
t = sp.symbols('t');
#xt, yt = sp.symbols('xt yt', cls=sp.Function);
xt = sp.Function("x")(t); # x(t)
yt = sp.Function("y")(t) # y(t)
g = f.subs([(x,xt),(y,yt)]);
gp = sp.diff(g,t);
print(g);
print(gp);
</code>
## Plotting symbolic functions
The sympy module has a `plot` method that knows how to plot symbolic functions of a single variable. The function `g` above with $x(t) = \sin(t)$ and $y(t) = \cos(2t)$ is a function of a single time variable `t`, and can be visualised as follows:
<code>
gs = g.subs([(xt,sp.sin(t)), (yt,sp.cos(2*t))]);
print(gs);
sp.plot(gs, (t,1,2));
</code>
A roughly equivalent plot could be obtained numerically by creating a lambda function for the expression, evaluating it for a closely-spaced set of values of `t` over the required range, and using standard numerical plotting functions that draw straight lines between the calulated points. If you increase the number of calculated points over the interval then the approximation in the above graph becomes more accurate.
<code>
import matplotlib.pyplot as plt
import numpy as np
%matplotlib notebook
tv = np.linspace(1, 2, 10);
gs_h = sp.lambdify(t, gs, modules=['numpy']);
gstv = gs_h(tv);
plt.plot(tv, gstv);
# lambda functions are also called anonymous functions. You can think of creating one as just another way of defining a function. For example:
# For example: say you had a symbolic expression, f = x^2
# writing my_func = sp.lambdify(x,f);
# is the same as going
# def my_func(x):
# x^2
</code>
The sympy plot function is quite fragile, and might not always work. Symbolic math packages are amazing, but they're difficult to implement and are sometimes not robust: you'll find various postings on the internet that give instances of very good symbolic math engines giving a wrong result. In short, they are useful but you should be careful when using them.
One other nice thing about Jupyter notebooks is that a pretty-print method exists for symbolic expressions. After the appropriate setup, note the difference in output between the `print` and `display` methods below:
<code>
from IPython.display import display
sp.init_printing() # pretty printing
print(gs);
display(gs);
</code>
## Symbolic integration
Integration is also a standard function in sympy, so we can find for example the integral
$$y(t) = \int_{-10}^t x(\lambda) d\lambda$$
for $x(t) = e^{-t/10} \cos(t)$:
<code>
xt = sp.exp(-t/10)*sp.cos(t); # x(t)
lamb = sp.symbols('lamb');
xl = xt.subs(t,lamb); # x(lamb)
yt = sp.integrate(xl, (lamb, -10, t)); # indefinite integral
yt
# to get a definite integral over the range, say -10 to 0, you'd go yt = sp.integrate(xl, (lamb, -10, 0));
# This would give a numeric value.
# NOTE: don't forget about your initial conditions. The definite integral just gives the change in the variable over the
# interval, so you need to add its initial state to this value get the true final state.
</code>
## Tasks
These tasks involve writing code, or modifying existing code, to meet the objectives described.
1. Define the expression $y(t) = v_0 t - \frac{1}{2} g t^2$ for some symbolic values of $v_0$ and $g$ using sympy. You should recognise this as the "altitude" of a particle moving under the influence of gravity, given that the initial velocity at time $t=0$ is $v_0$. Make a plot of the particle height in meters for $v_0 = 22.5m/s$ given $g = 9.8 m/s^2$, over the range $t=0$ to $t=5s$.<br><br>
2. Use symbolic math and the `roots` method to find an expression for the zeros of the expression $y(t)$ above for the same set of conditions. Substitute to find the nonzero numerical value of $t$ for which your plot in the previous task crosses the x-axis.<br><br>
For help on the `roots` method, type `?sp.roots()` in an empty code cell and run it. The method takes a symbolic expression as input, and returns a Python dictionary object as the output. The roots are contained in the tags of the dictionary. Note that for a d<br><br>
3. Use symbolic differentiation to find the vertical velocity of the particle in the previous task as a function of time, given the same conditions. Make a plot of this velocity over the same time range.<br><br>
4. Suppose the acceleration of a particle is given by $a(t) = 0.2 + \cos(t)$ for positive time. Use symbolic methods to find and plot the velocity $v(t)$ of the particle over the range $t=0$ to $t=5$ given the initial condition $v(0) = -0.3$. Then find and plot the position $s(t)$ of the particle over the same time period, given the additional auxiliary condition $s(0) = 0.1$.
<code>
?sp.roots
</code>
|
{
"filename": "lab_symintro_1.ipynb",
"repository": "nicollsf/eee2047s-notebooks",
"query": "transformed_from_existing",
"size": 127913,
"sha": ""
}
|
# scripts_Tabula_Muris_MM_2020.ipynb
Repository: xingjiepan/SCMG
<code>
%config InlineBackend.figure_format='retina'
</code>
<code>
import os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
from cytofuture_data.gene_name_mapping import GeneNameMapper
</code>
<code>
# Load the gene name mapper
gene_name_mapper = GeneNameMapper(
'../standard_genes/gene_names/human_genes.csv',
'../standard_genes/gene_names/mouse_genes.csv',
'../standard_genes/gene_names/orthologue_map_human2mouse_best.csv',
'../standard_genes/gene_names/orthologue_map_mouse2human_best.csv'
)
</code>
<code>
# Load the data
data_collection_name = 'Tabula_Muris_MM_2020'
data_path = os.path.join('/home/xingjie/Data/data2/cytofuture/datasets/scRNAseq/', data_collection_name)
dataset_name = '10x'
query_organism, query_var_id_type = 'mouse', 'id'
adata = sc.read_h5ad(os.path.join(data_path, 'download', 'adata_10x.h5ad'))
adata = adata.raw.to_adata()
adata
</code>
<code>
sc.pl.umap(adata, color='cell_type', palette='tab20')
</code>
<code>
sc.pp.calculate_qc_metrics(adata, percent_top=None, log1p=False, inplace=True)
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts'], jitter=0, multi_panel=True, log=True)
</code>
<code>
sc.pp.filter_cells(adata, min_genes=200)
</code>
<code>
# Append the dataset name into cel IDs
adata.obs.index = [':'.join([data_collection_name, dataset_name, i]) for i in adata.obs.index]
# Copy the standard meta-data
adata_std = sc.AnnData(X=adata.X, obs=adata.obs[[]], var=adata.var[[]])
adata_std.obs['dataset_id'] = ':'.join([data_collection_name, dataset_name])
adata_std.obs['assay'] = adata.obs['assay']
adata_std.obs['batch'] = [':'.join([data_collection_name, dataset_name, d])
for d in adata.obs['donor_id']]
adata_std.obs['development_stage'] = adata.obs['development_stage']
adata_std.obs['tissue'] = adata.obs['tissue']
adata_std.obs['cell_type'] = adata.obs['cell_type']
# Map the genes to human gene IDs
adata_std.var['human_gene_id'] = gene_name_mapper.map_gene_names(list(adata_std.var.index),
query_organism, 'human', query_var_id_type, 'id')
</code>
<code>
print(np.unique(adata_std.obs['tissue']))
print(np.unique(adata_std.obs['development_stage']))
display(adata_std)
display(adata_std[:, adata_std.var['human_gene_id'] != 'na'])
</code>
<code>
%%time
adata_std.write_h5ad(os.path.join(data_path, f'standard_adata_{data_collection_name}_{dataset_name}.h5ad'),
compression='gzip')
</code>
<code>
# Load the data
data_collection_name = 'Tabula_Muris_MM_2020'
data_path = os.path.join('/home/xingjie/Data/data2/cytofuture/datasets/scRNAseq/', data_collection_name)
dataset_name = 'smart-seq'
query_organism, query_var_id_type = 'mouse', 'id'
adata = sc.read_h5ad(os.path.join(data_path, 'download', 'adata_smart-seq.h5ad'))
adata = adata.raw.to_adata()
adata
</code>
<code>
sc.pl.umap(adata, color='cell_type', palette='tab20')
</code>
<code>
sc.pp.calculate_qc_metrics(adata, percent_top=None, log1p=False, inplace=True)
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts'], jitter=0, multi_panel=True, log=True)
</code>
<code>
sc.pp.filter_cells(adata, min_genes=200)
</code>
<code>
# Append the dataset name into cel IDs
adata.obs.index = [':'.join([data_collection_name, dataset_name, i]) for i in adata.obs.index]
# Copy the standard meta-data
adata_std = sc.AnnData(X=adata.X, obs=adata.obs[[]], var=adata.var[[]])
adata_std.obs['dataset_id'] = ':'.join([data_collection_name, dataset_name])
adata_std.obs['assay'] = adata.obs['assay']
adata_std.obs['batch'] = [':'.join([data_collection_name, dataset_name, d])
for d in adata.obs['donor_id']]
adata_std.obs['development_stage'] = adata.obs['development_stage']
adata_std.obs['tissue'] = adata.obs['tissue']
adata_std.obs['cell_type'] = adata.obs['cell_type']
# Map the genes to human gene IDs
adata_std.var['human_gene_id'] = gene_name_mapper.map_gene_names(list(adata_std.var.index),
query_organism, 'human', query_var_id_type, 'id')
</code>
<code>
print(np.unique(adata_std.obs['tissue']))
print(np.unique(adata_std.obs['development_stage']))
display(adata_std)
display(adata_std[:, adata_std.var['human_gene_id'] != 'na'])
</code>
<code>
%%time
adata_std.write_h5ad(os.path.join(data_path, f'standard_adata_{data_collection_name}_{dataset_name}.h5ad'),
compression='gzip')
</code>
|
{
"filename": "scripts_Tabula_Muris_MM_2020.ipynb",
"repository": "xingjiepan/SCMG",
"query": "transformed_from_existing",
"size": 8826,
"sha": ""
}
|
# RNA_ATAC_paired_scButterfly-C-checkpoint_1.ipynb
Repository: BioX-NKU/scButterfly
# RNA-ATAC: scButterfly-C
The following tutorial demonstrate how to use scButterfly-C variant with data augmentation using MultiVI cluster labels.
scButterfly-C with MultiVI cluster labels data augmentation will generate synthetic paired data by randomly paired scRNA-seq and scATAC-seq of different cells with the same cluster labels. [Leiden](https://www.nature.com/articles/s41598-019-41695-z/) is used on the embedding space of [MultiVI](https://www.nature.com/articles/s41592-023-01909-9), which is a single-cell multi-omics data joint analysis method in Python packages [scvi-tools](https://docs.scvi-tools.org/en/stable/). The supplement of these generated data will provide scButterfly a better performance of translation, but take more time for training.
<div class="alert note">
<p>
**Note**
Most of this tutorial is same as [scButterfly-B](RNA_ATAC_paired_scButterfly-B.ipynb) with more details of data pre-processing, model constructing, model training and evaluating. It's prefered to see that first, because it has no different in parts mentioned above, but have more useful notes.
</p>
</div>
<code>
from scButterfly.butterfly import Butterfly
import scanpy as sc
</code>
<code>
butterfly = Butterfly()
</code>
<code>
RNA_data = sc.read_h5ad('cellline_RNA_data.h5ad')
ATAC_data = sc.read_h5ad('cellline_ATAC_data.h5ad')
</code>
<code>
RNA_data
</code>
<code>
ATAC_data
</code>
<code>
from scButterfly.split_datasets import *
id_list = five_fold_split_dataset(RNA_data, ATAC_data)
train_id, validation_id, test_id = id_list[4]
</code>
<code>
butterfly.load_data(RNA_data, ATAC_data, train_id, test_id, validation_id)
</code>
<code>
butterfly.data_preprocessing()
</code>
<code>
butterfly.ATAC_data_p.var.chrom
</code>
<code>
chrom_list = []
last_one = ''
for i in range(len(butterfly.ATAC_data_p.var.chrom)):
temp = butterfly.ATAC_data_p.var.chrom[i]
if temp[0 : 3] == 'chr':
if not temp == last_one:
chrom_list.append(1)
last_one = temp
else:
chrom_list[-1] += 1
else:
chrom_list[-1] += 1
print(chrom_list, end="")
</code>
<code>
sum(chrom_list)
</code>
## Data augmentation with cluster labels from MultiVI embeddings
Here we choose the parameter ``aug_type="MultiVI_augmentation"``, and scButterfly-C will automatically train a MultiVI model first, and then generate randomly paired data as supplement for training.
<code>
butterfly.augmentation(aug_type="MultiVI_augmentation")
</code>
<code>
butterfly.construct_model(chrom_list=chrom_list)
</code>
Data augmentation will take more time for training.
<code>
butterfly.train_model()
</code>
<code>
A2R_predict, R2A_predict = butterfly.test_model()
</code>
<code>
from scButterfly.calculate_cluster import calculate_cluster_index
</code>
scButterfly-C usually get a better performance compare to [scButterfly-B](RNA_ATAC_paired_scButterfly-B.ipynb). However, the performance of MultiVI augmentation often slightly worse than data augmentation with cell-type labels. We recommend the [scButterfly-T](RNA_ATAC_paired_scButterfly-T.ipynb) when there has ``RNA_data.obs["cell_type"]`` and ``ATAC_data.obs["cell_type"]``.
<code>
sc.tl.tsne(A2R_predict)
sc.tl.leiden(A2R_predict)
sc.pl.tsne(A2R_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='small')
</code>
<code>
ARI, AMI, NMI, HOM = calculate_cluster_index(A2R_predict)
print('ATAC to RNA:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
</code>
<code>
sc.tl.tsne(R2A_predict)
sc.tl.leiden(R2A_predict)
sc.pl.tsne(R2A_predict, color=['cell_type', 'leiden'], legend_loc='on data', legend_fontsize='small')
</code>
<code>
ARI, AMI, NMI, HOM = calculate_cluster_index(R2A_predict)
print('RNA to ATAC:\nARI: %.3f, \tAMI: %.3f, \tNMI: %.3f, \tHOM: %.3f' % (ARI, AMI, NMI, HOM))
</code>
|
{
"filename": "RNA_ATAC_paired_scButterfly-C-checkpoint_1.ipynb",
"repository": "BioX-NKU/scButterfly",
"query": "transformed_from_existing",
"size": 140839,
"sha": ""
}
|
# index_1.ipynb
Repository: SheffieldML/notebook
## Sheffield ML Notebooks
This is a repository for the SheffieldML group's notebooks. They are broadly split into three categories.
* [Computational Biology and Bioinformatics](./compbio/index.ipynb) These notebooks are focussed on data analysis and new methodologies in computational biology.
* [GPy Examples](./GPy/index.ipynb) These notebooks give examples on how to use the group's GPy software.
* [GPy Philosophy](./GPy-phil/index.ipynb) These notebooks give examples on how to use the group's GPy software.
As well as these notebooks stored locally you can find examples of notebooks on our other github repositories.
### Open Data Science
For example [the ODS repo](https://github.com/sods/ods) has software for open data science including data set repositories. The notebooks can be found [here](https://github.com/sods/ods/blob/master/notebooks/pods/index.ipynb).
### Datasets
There are examples of the `pods` datasets avaiable [here](https://github.com/sods/ods/blob/master/notebooks/pods/datasets/index.ipynb).
* [Lab Classes for Teaching](./lab_classes/index.ipynb) These notebooks give lab classes for teaching including notebooks from Neil's Level 4/MSc module in machine learning.
|
{
"filename": "index_1.ipynb",
"repository": "SheffieldML/notebook",
"query": "transformed_from_existing",
"size": 2247,
"sha": ""
}
|
# 0.intro.ipynb
Repository: PCHN63101-Advanced-Data-Skills/R-Programming-Language
# Introduction
## Contents
```{tableofcontents}
```
## About the Authors
```{figure} images/george.jpg
---
scale: 80%
align: right
---
```
**Dr George Farmer | PhD**
Lecturer
... Dover Street Building | Division of Psychology, Communication and Human Neuroscience | School of Health Sciences | The University of Manchester M13 9PL
t: +44 (0)161 275 ... | e: [george.farmer@manchester.ac.uk](mailto:george.farmer@manchester.ac.uk)
```{figure} images/martyn.jpeg
---
scale: 15%
align: left
---
```
**Dr Martyn McFarquhar | PhD FHEA**
Lecturer in Neuroimaging
4.33 Booth Street East | Division of Psychology, Communication and Human Neuroscience | School of Health Sciences | The University of Manchester M13 9PL
t: +44 (0)161 275 2688 | e: [martyn.mcfarquhar@manchester.ac.uk](mailto:martyn.mcfarquhar@manchester.ac.uk)
|
{
"filename": "0.intro.ipynb",
"repository": "PCHN63101-Advanced-Data-Skills/R-Programming-Language",
"query": "transformed_from_existing",
"size": 2095,
"sha": ""
}
|
# speed_benchmark_3.ipynb
Repository: MaayanLab/blitzgsea
# Benchmark GSEA speed
Compare speed of GSEApy, fGSEA, and blitzGSEA. The runtime of fGSEA is calculated in a separate notebook as it runs in an R environment.
<code>
%%capture
!pip3 install git+https://github.com/MaayanLab/blitzgsea.git
</code>
<code>
!pip3 install gseapy
</code>
<code>
import importlib
import sys
sys.path.append('blitzgsea')
import blitzgsea as blitz
importlib.reload(blitz)
import gseapy
import time
import pandas as pd
import numpy as np
from matplotlib import pyplot as plt
import tqdm
</code>
<code>
!pip3 install gseapy --upgrade
</code>
<code>
import blitzgsea as blitz
import pandas as pd
# read signature as pandas dataframe
signature = pd.read_csv("https://github.com/MaayanLab/blitzgsea/raw/main/testing/ageing_muscle_gtex.tsv")
# list available gene set libraries in Enrichr
blitz.enrichr.print_libraries()
# use enrichr submodule to retrieve gene set library
library = blitz.enrichr.get_library("KEGG_2021_Human")
# run enrichment analysis
result = blitz.gsea(signature, library)
</code>
<code>
result
</code>
<code>
import pandas as pd
import blitzgsea as blitz
import gseapy
permutation_num = 1000
max_lib_size = 2000
outdir='testing/prerank_report_gobp'
format='png'
seed=1
processes = 1
signature = pd.read_csv("https://github.com/MaayanLab/blitzgsea/raw/main/testing/ageing_muscle_gtex.tsv")
library = blitz.enrichr.get_library("GO_Biological_Process_2021")
sig = signature.sort_values(1, ascending=False)
sig = signature[~signature.index.duplicated(keep='first')]
pre_res = gseapy.prerank(rnk=sig, gene_sets=library, processes=processes, permutation_num=permutation_num, outdir=outdir, format=format, seed=seed)
</code>
<code>
signature
</code>
<code>
signature = pd.read_csv("testing/ageing_muscle_gtex.tsv")
library = blitz.enrichr.get_library("GO_Biological_Process_2021")
</code>
## blitzGSEA benchmark
Calculate runtime for 6000 gene sets for different number of threads with signature cache turned off. The signature cache would avoid recalculating anchor parameters. Run blitzGSEA with signature cache activated ones. Only the probabilies will be caluclated. This step does not benefit from multi-threading as it is memory constraint.
<code>
tt = []
for i in tqdm.tqdm(range(8,9)):
st = time.time()
blitz.gsea(signature, library, processes=i, permutations=4000, min_size=5, max_size=4000, signature_cache=False, center=False)
tt.append(time.time()-st)
print(tt)
st = time.time()
res = blitz.gsea(signature, library, min_size=5, max_size=4000, center=False, deep_accuracy=200)
sigcache = time.time()-st
</code>
<code>
sigh = [sigcache]*len(tt)
sigh
</code>
<code>
term = "ubiquitin-dependent protein catabolic process (GO:0006511)"
print(res.loc[term, :])
fig = blitz.plot.running_sum(signature, term, library, result=res, compact=False, center=False)
plt.show()
</code>
## Compute GSEApy runtime
Library needs to be chunked due to very high memory requirements. Chunking does not slow down the algorithm significanltly.
<code>
permutation_num = 1000
max_lib_size = 100
outdir='testing/prerank_report_kegg'
format='png'
seed=1
processes= 3
gpy = []
signature.columns = ["a", "b"]
sig = signature.sort_values("b", ascending=False)
sig.set_index("a")
sig = signature[~signature.index.duplicated(keep='first')]
def chopped_gsea(rnk, gene_sets, processes, permutation_num=1000, max_lib_size=50, outdir='test/prerank_report_kegg', format='png', seed=1):
library_keys = list(gene_sets.keys())
chunks = [library_keys[i:i+max_lib_size] for i in range(0, len(library_keys), max_lib_size)]
results = []
for chunk in chunks:
tlib = {}
for k in chunk:
tlib[k] = gene_sets[k]
pre_res = gseapy.prerank(rnk=rnk, gene_sets=tlib, processes=processes, permutation_num=permutation_num, outdir=outdir, format=format, seed=seed)
results.append(pre_res.res2d)
return pd.concat(results)
tt_gseapy = []
for p in range(1,9):
st = time.time()
res2 = chopped_gsea(sig, library, p, permutation_num=permutation_num, max_lib_size=50)
et2 = time.time() - st
tt_gseapy.append(et2)
</code>
<code>
import time
permutation_num = 2000
max_lib_size = 100
outdir='testing/prerank_report_kegg'
#signature = pd.read_csv("testing/ageing_muscle_gtex.tsv")
signature = pd.read_csv("https://github.com/MaayanLab/blitzgsea/raw/main/testing/ageing_muscle_gtex.tsv")
#library = blitz.enrichr.get_library("KEGG_2021_Human")
library = blitz.enrichr.get_library("GO_Biological_Process_2021")
st = time.time()
blitz_res = blitz.gsea(signature, library, min_size=5, max_size=4000, permutations=permutation_num, center=False, deep_accuracy=200, signature_cache=False)
print("blitz:", time.time()-st)
st = time.time()
blitz_res = blitz.gsea(signature, library, min_size=5, max_size=4000, permutations=permutation_num, center=False, deep_accuracy=200, signature_cache=True)
print("blitz cache:", time.time()-st)
#signature = pd.read_csv("testing/ageing_muscle_gtex.tsv")
signature = pd.read_csv("https://github.com/MaayanLab/blitzgsea/raw/main/testing/ageing_muscle_gtex.tsv")
signature.columns = ["i", "v"]
sig = signature.sort_values("v", ascending=False)
sig = signature[~signature.index.duplicated(keep='first')]
st = time.time()
gseapy.prerank(rnk=sig, gene_sets=library, outdir=outdir, permutation_num=permutation_num, seed=1337)
print("gseapy:", time.time()-st)
gseapy_res = pd.read_csv(outdir+"/gseapy.gene_set.prerank.report.csv", sep=",")
gseapy_res.index = gseapy_res.loc[:,"Term"]
</code>
<code>
import matplotlib.pyplot as plt
from matplotlib.patches import Patch
data = [66.94, 12.9, 6.24, 1.99]
labels = ["GSEApy", "fGSEA", "blitzGSEA", "blitzGSEA (cache)"]
colors = ['blue', 'orange', 'green', 'red']
plt.bar(range(len(data)), data, color=colors)
plt.xlabel("Methods")
plt.ylabel("runtime (sec)")
plt.title("Runtime for GO: Biological Process Enrichment")
plt.xticks(range(len(data)), labels)
legend_patches = [Patch(color=color) for color in colors]
plt.legend(legend_patches, labels)
plt.show()
</code>
<code>
from matplotlib import pyplot as plt
import numpy as np
inter = blitz_res.index.intersection(gseapy_res.index)
bp = -np.log(blitz_res.loc[inter, "pval"])
gp = -np.log(gseapy_res.loc[inter, "NOM p-val"])
max_val = np.max(gp[np.isfinite(gp)])
gp[gp == np.inf] = max_val
plt.scatter(bp, gp)
x_line = np.array([0, 8])
y_line = x_line
# Plot the line
plt.plot(x_line, y_line, 'r--')
plt.xlabel("blitzgsea -log(p-val)", fontsize=16)
plt.ylabel("GSEApy -log(p-val)", fontsize=16)
</code>
<code>
permutation_num = 2000
max_lib_size = 100
outdir='testing/prerank_report_kegg'
signature = pd.read_csv("testing/ageing_muscle_gtex.tsv")
library = blitz.enrichr.get_library("KEGG_2021_Human")
st = time.time()
blitz_res_1000 = blitz.gsea(signature, library, min_size=5, max_size=4000, permutations=permutation_num, center=False, deep_accuracy=200, signature_cache=False)
print("blitz:", time.time()-st)
signature = pd.read_csv("testing/ageing_muscle_gtex.tsv")
signature.columns = ["i", "v"]
sig = signature.sort_values("v", ascending=False)
sig = signature[~signature.index.duplicated(keep='first')]
st = time.time()
gseapy.prerank(rnk=sig, gene_sets=library, outdir=outdir, permutation_num=permutation_num, seed=1337)
print("gseapy:", time.time()-st)
gseapy_res_1000 = pd.read_csv(outdir+"/gseapy.gene_set.prerank.report.csv", sep=",")
gseapy_res_1000.index = gseapy_res_1000.loc[:,"Term"]
</code>
<code>
from matplotlib import pyplot as plt
import numpy as np
inter = gseapy_res_1000.index.intersection(gseapy_res.index)
bp = -np.log(gseapy_res_1000.loc[inter, "NOM p-val"])
gp = -np.log(gseapy_res.loc[inter, "NOM p-val"])
max_val = np.max(gp[np.isfinite(gp)])
gp[gp == np.inf] = max_val
max_val = np.max(bp[np.isfinite(bp)])
bp[bp == np.inf] = max_val
plt.scatter(bp, gp)
x_line = np.array([0, 7])
y_line = x_line
# Plot the line
plt.plot(x_line, y_line, 'r--')
plt.xlabel("GSEApy 1000 -log(p-val)", fontsize=16)
plt.ylabel("GSEApy 6000 -log(p-val)", fontsize=16)
</code>
<code>
from matplotlib import pyplot as plt
import numpy as np
inter = blitz_res_1000.index.intersection(gseapy_res.index)
bp = -np.log(blitz_res_1000.loc[inter, "pval"])
gp = -np.log(blitz_res.loc[inter, "pval"])
plt.scatter(bp, gp)
x_line = np.array([0, 12])
y_line = x_line
# Plot the line
plt.plot(x_line, y_line, 'r--')
plt.xlabel("blitzGSEA 1000 -log(p-val)", fontsize=16)
plt.ylabel("blitzGSEA 6000 -log(p-val)", fontsize=16)
</code>
## Plot results
Plot the results for GSEApy, fGSEA, and blitzGSEA. Speed for GSEApy and fGSEA was precomputed. fGSEA does not support multi-threading and the average runtime is plotted as a constant.
<code>
width = 0.5
labels = range(1,len(tt)+1)
# 12.7578 computed in R
fgsea = [12.7578]*len(tt)
x = np.array(range(1,len(tt)+1))*2
xt = np.array(x)
xt[0] = 3
tt_gseapy = tt_gseapy[0:len(tt)]
f, (ax, ax2) = plt.subplots(2, 1, sharex=True)
rects1 = ax.bar(xt-0.25, tt, width, label='blitzGSEA (anchor)')
rects2 = ax.bar(xt-0.25, sigh, width, label='blitzGSEA (prob)')
rects3 = ax.bar(2, 12.7578, width, label='fGSEA')
rects4 = ax.bar(2-0.5, 1830.63, width, label='GSEApy')
#rects5 = ax.bar(xt+0.25, tt_gseapy, width, label='GSEApy')
rects1 = ax2.bar(xt-[0.5,0,0,0,0,0,0], tt, width, label='blitzGSEA (anchor)')
rects2 = ax2.bar(xt-[0.5,0,0,0,0,0,0], sigh, width, label='blitzGSEA (prob)')
rects3 = ax2.bar(2, 12.7578, width, label='fGSEA')
rects4 = ax2.bar(2-0.5, 200, width, label='GSEApy')
d = .015 # how big to make the diagonal lines in axes coordinates
# arguments to pass to plot, just so we don't keep repeating them
kwargs = dict(transform=ax.transAxes, color='k', clip_on=False)
ax.plot((-d, +d), (-d, +d), **kwargs) # top-left diagonal
ax.plot((1 - d, 1 + d), (-d, +d), **kwargs) # top-right diagonal
ax.tick_params(labelsize=16)
kwargs.update(transform=ax2.transAxes) # switch to the bottom axes
ax2.plot((-d, +d), (1 - d, 1 + d), **kwargs) # bottom-left diagonal
ax2.plot((1 - d, 1 + d), (1 - d, 1 + d), **kwargs) # bottom-right diagonal
ax.set_ylim(min(tt_gseapy)-50, max(tt_gseapy)+50) # outliers only
ax2.set_ylim(0, 25) # most of the data
ax.spines['bottom'].set_visible(False)
ax2.spines['top'].set_visible(False)
ax.xaxis.tick_top()
ax.tick_params(labeltop=False) # don't put tick labels at the top
ax2.xaxis.tick_bottom()
ax.set_xticks(x)
ax.set_xticklabels(labels)
ax2.set_ylabel('seconds', fontsize=20)
ax2.set_xlabel('threads', fontsize=20)
plt.tick_params(labelsize=16)
ax.legend(fontsize=16, frameon=False)
plt.savefig("figures/speed_threads_fgsea.pdf", bbox_inches='tight')
</code>
|
{
"filename": "speed_benchmark_3.ipynb",
"repository": "MaayanLab/blitzgsea",
"query": "transformed_from_existing",
"size": 334745,
"sha": ""
}
|
# association.ipynb
Repository: tmichoel/BioFindrTutorials
## Introduction
While [BioFindr][1] is developed primarily for causal inference from genomics and transcriptomics data, association analysis between genomics and transcriptomics data is also possible. In association analysis, genetic effects on the transcriptome are measured by testing if genes are differentially expressed in different groups of samples defined by the genotype of a genetic variant of interest. In [BioFindr][1], significance of association is computed using a [categorical model](https://tmichoel.github.io/BioFindr.jl/dev/realLLR/#prim_test_realLLR) and a variant-specific background distribution. Similar to what was done in the [coexpression analysis tutorial](coexpression.qmd), this is achieved by modelling the distribution of association values between a given variant $A$ and all genes $B$ as a [mixture distribution](https://en.wikipedia.org/wiki/Mixture_distribution) of real and null (random) associations. The relative weight of each component then reflects the prior probability of finding a non-null $B$ gene for a given variant $A$, and is fitted for every $A$ separately.
We will illustrate how to run association analysis with [BioFindr][1] using [preprocessed data][2] from the [GEUVADIS study][3]. See the [installation instructions](installation.qmd) for the steps you need to take to reproduce this tutorial.
## Set up the environment
We begin by setting up the environment and loading some necessary packages.
<code>
using DrWatson
quickactivate(@__DIR__)
using DataFrames
using Arrow
using BioFindr
</code>
## Load data
### Expression data
[BioFindr][1] expects that expression data are stored as [floating-point numbers](https://docs.julialang.org/en/v1/manual/integers-and-floating-point-numbers/) in a [DataFrame][4] where columns correspond to variables (genes) and rows to samples, see the [coexpression analysis tutorial](coexpression.qmd) for more details.
This tutorial uses two tables of expression data from the same set of samples, one for mRNA expression data called `dt`, and one for microRNA (miRNA) expression data called `dm`:
<code>
dt = DataFrame(Arrow.Table(datadir("exp_pro","findr-data-geuvadis", "dt.arrow")));
dm = DataFrame(Arrow.Table(datadir("exp_pro","findr-data-geuvadis", "dm.arrow")));
</code>
### Genotype data
[BioFindr][1] expects that genotype data are stored as [integer numbers](https://docs.julialang.org/en/v1/manual/integers-and-floating-point-numbers/) in a [DataFrame][4] where columns correspond to variables (genetic variants) and rows to samples. Since [BioFindr][1] uses a [categorical association model](https://tmichoel.github.io/BioFindr.jl/dev/realLLR/#prim_test_realLLR), it does not matter how different genotypes (e.g. heterozygous vs. homozygous) are encoded as integers. Future versions will support [scientific types](https://juliaai.github.io/ScientificTypes.jl/dev/) for representing genotype data.
This tutorial uses two tables of genotype data from the same set of samples as the expression data, one with genotypes for mRNA eQTLs called `dgt`, and one for microRNA (miRNA) eQTLs called `dgm`:
<code>
dgt = DataFrame(Arrow.Table(datadir("exp_pro","findr-data-geuvadis", "dgt.arrow")));
dgm = DataFrame(Arrow.Table(datadir("exp_pro","findr-data-geuvadis", "dgm.arrow")));
</code>
## Run BioFindr
Assume we are interested in identifying mRNA genes whose expression levels are associated to microRNA eQTLs. We run:
<code>
dP = findr(dt, dgm, FDR=0.05)
</code>
BioFindr computes a [posterior probability](https://tmichoel.github.io/BioFindr.jl/dev/posteriorprobs/) of non-zero association for every **Source** variant (columns of `dgm`) and **Target** gene (columns of `dt`). By default the output is sorted by decreasing **Probability**. The optional parameter **FDR** can be used to limit the output to the set of pairs that has a [global false discovery rate (FDR)](https://en.wikipedia.org/wiki/False_discovery_rate#Storey-Tibshirani_procedure) less than a desired value (here set to 5%). The **qvalue** column in the output can be used for further filtering of the output, see the [coexpression analysis tutorial](coexpression.qmd) for further details.
Note the order of the arguments. The first argument `dt` is the **Target** DataFrame, and the second argument the **Source** DataFrame.
[1]: https://github.com/tmichoel/BioFindr.jl
[2]: https://github.com/lingfeiwang/findr-data-geuvadis
[3]: https://doi.org/10.1038/nature12531
[4]: https://dataframes.juliadata.org/stable/
[5]: https://doi.org/10.1371/journal.pcbi.1005703
|
{
"filename": "association.ipynb",
"repository": "tmichoel/BioFindrTutorials",
"query": "transformed_from_existing",
"size": 6848,
"sha": ""
}
|
# P6_2019_web_nn_101.ipynb
Repository: krzakala/ml
# Classification sur MNIST avec un reseau de neuronnes
<code>
import numpy as np
import matplotlib.pyplot as plt
import keras
from keras.datasets import mnist
from keras.models import Sequential
from keras.layers import Dense
from keras.optimizers import RMSprop
%matplotlib inline
</code>
Nous commençons par charger les données MNIST. À la différence d’avant, nous n’utiliserons pas scikitlearn pour le faire, mais la bibliothèque Keras. C'est parce que nous allons passer des méthodes "classiques" à ce que l'on appelle "l'apprentissage en profondeur", c'est-à-dire l'apprentissage automatique à l'aide de réseaux de neurones profonds.
Keras s’appuie sur Tensorflow, un framework d’apprentissage en profondeur développé par Google et probablement le plus utilisé par les praticiens (PyTorch, développé sur Facebook, est une alternative qui gagne du terrain.)
<code>
# Download the data, shuffled and split between train and test sets
(X_train, y_train), (X_test, y_test) = mnist.load_data()
print("no. of training/test samples: %d, %d" % (X_train.shape[0], X_test.shape[0]))
print("no. of features: %d" % (np.product(X_train.shape[1:])))
# Let us look at some of the digits
fig, axs = plt.subplots(2, 2)
for d in range(4):
axs[d % 2, d // 2].imshow(X_train[d], cmap="gray")
axs[d % 2, d // 2].set_title("correct label: %d" % (y_train[d]))
fig.tight_layout()
</code>
Notez que Keras charge MNIST de manière légèrement différente de scikitlearn: les données sont déjà mélangées et divisées dans des ensembles d’entraînement / test; de plus, chaque échantillon est un tableau 2D de dimension 28x28, au lieu d’un tableau 1D de dimension 784.
Nous procédons par un prétraitement. Nous allons * rasteriser * ces vecteurs, c'est-à-dire les transformer de tableaux bidimensionnels en tableaux unidimensionnels; de plus, nous les normaliserons, de sorte que chaque composant du tableau ait une valeur comprise entre 0 et 1, au lieu de 0 et 255.
<code>
# Rasterize and normalize samples
X_train = X_train.reshape(X_train.shape[0], -1)
X_test = X_test.reshape(X_test.shape[0], -1)
X_train = X_train / 255
X_test = X_test / 255
# Use 32-bit instead of 64-bit float
X_train = X_train.astype("float32")
X_test = X_test.astype("float32")
# Work with one-hot encoding of labels
n_classes = len(np.unique(y_train))
y_train_b = keras.utils.to_categorical(y_train, n_classes)
y_test_b = keras.utils.to_categorical(y_test, n_classes)
print("The categories of the images above is now encoded as: \n", y_train_b[0:4])
</code>
Il est maintenant temps de construire notre réseau de neurones multicouches. Nous allons d'abord le faire avec une seule couche cachée.
Dans le code ci-dessous, nous pouvons voir à quel point il est facile de configurer des modèles dans Keras. En quelques lignes, nous avons construit un réseau de neurones à 2 couches, et généraliser à partir de celle-ci en plusieurs couches est assez simple.
<code>
# Specify model
model = Sequential()
model.add(Dense(512, activation="relu", input_shape=(784,)))
model.add(Dense(n_classes, activation="softmax"))
# Print model and compile it
model.summary()
model.compile(loss="categorical_crossentropy",
optimizer=RMSprop(),
metrics=["accuracy"])
</code>
Notez que la première couche dense a $ 784 \times 512 + 512 $ paramètres, la seconde en a $ 512 \times 10 + 10 $. Ils correspondent aux poids, plus un biais appliqué à chaque sortie. Nous compilons le modèle en utilisant l'entropie croisée comme fonction de cout (l'equivalent de la regression logistique) et nous utiliserons l'algorithme d'optimisation RMSprop décrit [ici] (http://ruder.io/optimizing-gradient-descent/index.html#rmsprop) ainsi que d'autres variantes de descente de gradient. .
Après avoir configuré le modèle, nous pouvons l'entrainer comme dans scikitlearn: en utilisant `model.fit (X_train, y_train)`.
<code>
# Parameters
batch_size = 128
epochs = 20
# Perform fit
history = model.fit(X_train, y_train_b,
batch_size=batch_size,
epochs=epochs,
verbose=1,
shuffle=False,
validation_data=(X_test, y_test_b))
# Print results
score = model.evaluate(X_test, y_test_b, verbose=0)
print('Test loss/accuracy: %g, %g' % (score[0], score[1]))
</code>
<code>
plt.figure(figsize=(15, 5))
# Plot history for accuracy
plt.subplot(121)
plt.plot(history.history['acc'])
plt.plot(history.history['val_acc'])
plt.title('model accuracy -- MLP')
plt.ylabel('accuracy')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
# summarize history for loss
plt.subplot(122)
plt.plot(history.history['loss'])
plt.plot(history.history['val_loss'])
plt.title('model loss -- MLP')
plt.ylabel('loss')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
plt.tight_layout()
</code>
C’est en fait assez bon, une précision de 2% representait l’état de l'art pendant une longue periode (vérifiez-le [ici] (http://yann.lecun.com/exdb/mnist/)), meme si l'on peut faire bien mieux maintenant, comme nous le verrons plus tard. Juste par curiosité, vous trouverez [ici] (http://rodrigob.github.io/are_we_there_yet/build/classification_datasets_results.html) est une liste détaillée des meilleurs résultats que les utilisateurs ont pu obtenir sur MNIST. La plus petite erreur de test obtenue jusqu’à présent est de 0,21%, c’est-à-dire que seulement 21 des 10 000 échantillons de test ne sont pas classés correctement!
## Jouer avec notre reseau de neuronnes
Il existe de nombreux degrés de liberté dans notre modèle et dans la procédure de formation, qui ont été choisis de manière plus ou moins arbitraire. Par exemple, pourquoi utilisons-nous ces valeurs pour 'batch_size' et 'epochs'? Pourquoi utilisons-nous une activation ReLU sur la 1ère couche et une activation softmax sur la seconde? Pourquoi utilisons-nous cette perte, cet optimiseur, etc.
C’est l’un des principaux problèmes de l’apprentissage en profondeur: en principe, on ne sait pas quelle approche donnera les meilleurs résultats, et de nombreuses approches sont possibles. Il faut donc essayer beaucoup de choses et voir ce qui se passe.
Nous pouvons procéder à une validation croisée, mais les possibilités sont tellement nombreuses que tout essayer n'est tout simplement pas réalisable.
Nous avons donc besoin de développer un instinct sur ce qui convient le mieux à chaque situation - parfois, la théorie peut nous guider, parfois non.
Nous allons maintenant voir comment les choses changent si nous ajoutons l'abandon et la normalisation des lots, prenons un réseau plus superficiel ou ajoutons une deuxième couche. Il semble toutefois difficile de trop améliorer la performance de 2% avec cette approche.
### dropout & batch normalization
En regardant les chiffres imprimés à l'écran pendant la formation, on s'aperçoit que notre modèle surajustait beaucoup: nous obtenons essentiellement une précision de 100% sur l'ensemble de formation!
Certaines astuces ont été introduites pour gérer ce surajustement. Le premier d'entre eux s'appelle * dropout * (introduit [ici] (https://www.cs.toronto.edu/~hinton/absps/JMLRdropout.pdf)), et consiste, à chaque étape de la procédure de formation , en ignorant certaines des fonctionnalités. C’est plus ou moins ce que nous avons fait lorsque nous avons examiné des forêts aléatoires: chacun des arbres de la forêt utilisait un sous-ensemble de caractéristiques. On peut donc voir le décrochage comme une sorte de procédure d’ensachage qui rend le modèle plus robuste. La différence est que la suppression peut être appliquée à chaque couche, pas seulement à la première couche en entrée.
L’autre astuce est la * normalisation par batch *, qui consiste à prélever chaque échantillon dans un mini-lot et à le standardiser à l’aide de la moyenne et de la variance * mini-batch *, voir [ici] (https://arxiv.org/pdf/1502.03167 .pdf) pour plus de détails.
Ces astuces ont été introduites principalement de manière ad-hoc - les gens ont quelques explications sur les raisons pour lesquelles ils fonctionnent et ont essayé de faire de la théorie à ce sujet, mais au final, nous ne comprenons toujours pas vraiment pourquoi et quand ils fonctionnent ( voir [le discours provocateur d'Ali Rahimi] (https://www.youtube.com/watch?v=Qi1Yry33TQE)).
<code>
from keras.layers import Dropout, BatchNormalization, Activation
# Parameters
batch_size = 128
epochs = 20
# Specify model
model = Sequential()
model.add(Dense(512, input_dim=784, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("relu"))
model.add(Dropout(0.2))
model.add(Dense(n_classes, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("softmax"))
# Print model and compile it
model.summary()
model.compile(loss="categorical_crossentropy",
optimizer=RMSprop(),
metrics=["accuracy"])
# Perform fit
history_dropout = model.fit(X_train, y_train_b,
batch_size=batch_size,
epochs=epochs,
verbose=1,
validation_data=(X_test, y_test_b))
# Print results
score = model.evaluate(X_test, y_test_b, verbose=0)
print('Test loss/accuracy: %g, %g' % (score[0], score[1]))
</code>
<code>
plt.figure(figsize=(15, 5))
# Plot history for accuracy
plt.subplot(121)
plt.plot(history_dropout.history['acc'])
plt.plot(history_dropout.history['val_acc'])
plt.title('model accuracy -- MLP with dropout and BN')
plt.ylabel('accuracy')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
# summarize history for loss
plt.subplot(122)
plt.plot(history_dropout.history['loss'])
plt.plot(history_dropout.history['val_loss'])
plt.title('model loss -- MLP with dropout and BN')
plt.ylabel('loss')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
plt.tight_layout()
</code>
### D'autre algorithmes d'optimisation
Nous utilisons un optimiseur appelé RMSprop, qui consiste en une descente de gradient avec quelques termes supplémentaires impliqués. Il y a cependant d'autres possibilités. Adam, un autre optimiseur basé sur la descente de gradient, a récemment été introduit et a montré de très bons résultats en pratique.
De bonnes comparaisons de différents optimiseurs sont disponibles [ici] (http://ruder.io/optimizing-gradient-descent/) et [ici] (https://3dbabove.com/2017/11/14/optimizationalgorithms/).
<code>
from keras.optimizers import adam
# Parameters
batch_size = 128
epochs = 20
# Specify model
model = Sequential()
model.add(Dense(512, input_dim=784, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("relu"))
model.add(Dropout(0.2))
model.add(Dense(n_classes, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("softmax"))
# Print model and compile it
model.summary()
model.compile(loss="categorical_crossentropy",
optimizer=adam(),
metrics=["accuracy"])
# Perform fit
history_adam = model.fit(X_train, y_train_b,
batch_size=batch_size,
epochs=epochs,
verbose=1,
validation_data=(X_test, y_test_b))
# Print results
score = model.evaluate(X_test, y_test_b, verbose=0)
print('Test loss/accuracy: %g, %g' % (score[0], score[1]))
</code>
<code>
plt.figure(figsize=(15, 5))
# Plot history for accuracy
plt.subplot(121)
plt.plot(history_adam.history['acc'])
plt.plot(history_adam.history['val_acc'])
plt.title('model accuracy -- MLP using Adam')
plt.ylabel('accuracy')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
# summarize history for loss
plt.subplot(122)
plt.plot(history_adam.history['loss'])
plt.plot(history_adam.history['val_loss'])
plt.title('model loss -- MLP using Adam')
plt.ylabel('loss')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
plt.tight_layout()
</code>
### Rajouter des couches...
Et si nous ajoutions simplement une autre couche? Disons que c'est identique au premier (bien que cela puisse être autre chose - les possibilités ici sont infinies!)
<code>
# Parameters
batch_size = 128
epochs = 20
# Specify model
model = Sequential()
model.add(Dense(512, input_dim=784, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("relu"))
model.add(Dropout(0.2))
model.add(Dense(512, input_dim=784, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("relu"))
model.add(Dropout(0.2))
model.add(Dense(n_classes, init="uniform"))
model.add(BatchNormalization())
model.add(Activation("softmax"))
# Print model and compile it
model.summary()
model.compile(loss="categorical_crossentropy",
optimizer=adam(),
metrics=["accuracy"])
# Perform fit
history_onemorelayer = model.fit(X_train, y_train_b,
batch_size=batch_size,
epochs=epochs,
verbose=1,
shuffle=False,
validation_data=(X_test, y_test_b))
# Print results
score = model.evaluate(X_test, y_test_b, verbose=0)
print('Test loss/accuracy: %g, %g' % (score[0], score[1]))
</code>
<code>
plt.figure(figsize=(15, 5))
# Plot history for accuracy
plt.subplot(121)
plt.plot(history_onemorelayer.history['acc'])
plt.plot(history_onemorelayer.history['val_acc'])
plt.title('model accuracy -- MLP with three layers')
plt.ylabel('accuracy')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
# summarize history for loss
plt.subplot(122)
plt.plot(history_onemorelayer.history['loss'])
plt.plot(history_onemorelayer.history['val_loss'])
plt.title('model loss -- MLP with three layers')
plt.ylabel('loss')
plt.xlabel('epoch')
plt.legend(['train', 'test'], loc='upper left')
plt.tight_layout()
</code>
Comme nous pouvons le constater, il est très difficile de dépasser la précision de test de 98%. Pour ce faire, nous devrons examiner des modèles encore plus compliqués - plus précisément, des couches autres que celle dense. Nous ferons cela lors du prochain cours!
|
{
"filename": "P6_2019_web_nn_101.ipynb",
"repository": "krzakala/ml",
"query": "transformed_from_existing",
"size": 135978,
"sha": ""
}
|
# idea_Immunology1_1.ipynb
Repository: Elizaluckianchikova/Bioinformatics
<a href="https://colab.research.google.com/github/Elizaluckianchikova/Bioinformatics_idea/blob/main/Immunology1.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a>
**Моделиронивание силы иммунного ответа на инфекцию **
Моделирование силы иммунного ответа в Python может быть выполнено с использованием различных методов и библиотек, таких как моделирование системы дифференциальных уравнений, агентно-ориентированное моделирование или машинное обучение.
Одним из подходов к моделированию иммунного ответа является использование моделей дифференциальных уравнений, которые описывают динамику взаимодействия между различными клетками и сигнальными молекулами в иммунной системе. Для реализации таких моделей в Python можно воспользоваться библиотекой SciPy, которая предоставляет инструменты для численного решения дифференциальных уравнений.
В этом примере мы определяем функцию immune_response, которая содержит систему дифференциальных уравнений, описывающих взаимодействие между иммунными клетками и вирусами. Мы затем используем функцию solve_ivp из SciPy для численного решения этой системы уравнений и визуализации результатов.
Это простой пример моделирования иммунного ответа. В зависимости от конкретных потребностей и целей моделирования, можно создать более сложные модели и учитывать различные аспекты иммунной системы. Если у вас есть конкретные вопросы или требуется дальнейшее пояснение, не стесняйтесь задавать их.
<code>
import numpy as np
from scipy.integrate import solve_ivp
import matplotlib.pyplot as plt
# Определение функции, описывающей систему дифференциальных уравнений для модели иммунного ответа
def immune_response(t, y):
# Параметры модели
k1 = 0.1
k2 = 0.05
# Уравнения модели
dTdt = -k1 * y[0] # Иммунные клетки
dVdt = k1 * y[0] - k2 * y[1] # Вирусы
return [dTdt, dVdt]
# Начальные условия
y0 = [100, 10] # Иммунные клетки и вирусы
# Временной интервал моделирования
t_span = (0, 10)
# Решение системы дифференциальных уравнений
sol = solve_ivp(immune_response, t_span, y0, t_eval=np.linspace(0, 10, 100))
# Визуализация результатов
plt.figure()
plt.plot(sol.t, sol.y[0], label='Иммунные клетки')
plt.plot(sol.t, sol.y[1], label='Вирусы')
plt.xlabel('Время')
plt.ylabel('Количество')
plt.legend()
plt.show()
</code>
|
{
"filename": "idea_Immunology1_1.ipynb",
"repository": "Elizaluckianchikova/Bioinformatics",
"query": "transformed_from_existing",
"size": 38993,
"sha": ""
}
|
# Base-02-GRN_preparation_for_CellOracle.ipynb
Repository: tmnolan/Brassinosteroid-gene-regulatory-networks-at-cellular-resolution
<code>
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
%matplotlib inline
import seaborn as sns
import os, sys, shutil, importlib, glob
from tqdm.notebook import tqdm
from celloracle import motif_analysis as ma
</code>
<code>
print(pd.__version__)
</code>
<code>
print(np.__version__)
</code>
<code>
%config InlineBackend.figure_format = 'retina'
plt.rcParams['figure.figsize'] = [6, 4.5]
plt.rcParams["savefig.dpi"] = 300
</code>
<code>
os.chdir('/fast/AG_Ohler/CheWei/Root_scATAC/cicero_output')
</code>
<code>
# Load scATAC-seq peak list.
peaks = pd.read_csv("./all_peaks.csv", index_col=0)
peaks = peaks.x.values
peaks
</code>
<code>
# Load cicero coaccess score.
cicero_connections = pd.read_csv("cicero_connections.csv", index_col=0)
cicero_connections.head()
</code>
<code>
## Make TSS annotation
ma.SUPPORTED_REF_GENOME
</code>
<code>
tss_annotated = ma.get_tss_info(peak_str_list=peaks, ref_genome="TAIR10") ##!! Set reference genome here
# Check results
tss_annotated.tail()
</code>
<code>
tss_annotated['chr'] = tss_annotated['chr'].astype(str)
</code>
<code>
## Integrate TSS info into cicero connection
integrated = ma.integrate_tss_peak_with_cicero(tss_peak=tss_annotated, cicero_connections=cicero_connections)
print(integrated.shape)
integrated.head()
</code>
<code>
## Filter peaks
peak = integrated[integrated.coaccess >= 0.8]
peak = peak[["peak_id", "gene_short_name"]].reset_index(drop=True)
</code>
<code>
print(peak.shape)
peak.head()
</code>
<code>
peak.to_csv("processed_peak_file.csv")
</code>
|
{
"filename": "Base-02-GRN_preparation_for_CellOracle.ipynb",
"repository": "tmnolan/Brassinosteroid-gene-regulatory-networks-at-cellular-resolution",
"query": "transformed_from_existing",
"size": 18617,
"sha": ""
}
|
# lauzhacktest.ipynb
Repository: ihchaeryu/LauzHack23-RHR
<code>
pip install openai==0.28
</code>
<code>
pip install tiktoken
</code>
<code>
import numpy as np
import scipy
import matplotlib.pyplot as plt
import openai
import pandas as pd
import tiktoken
</code>
<code>
openai.api_key = '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'
</code>
<code>
def generate_response(prompt):
messag=[{"role": "system", "content": "You are a chatbot"}]
## build a chat history: you can CONDITION the bot on the style of replies you want to see - also getting weird behaviors... such as KanyeGPT
history_bot = ["Yes, I'm ready! Please provide the first paper abstract."]
# ask ChatGPT to return STRUCTURED, parsable answers that you can extract easily - often better providing examples of desired behavior (1-2 example often enough)
history_user = ["i'll give you some paper abstracts. for each abstract (i.e., for each of my messages), you will a) assign a topic from the following list:\nbiochemistry\nbiophysics\nproteomics\ncancer\ncell biology\nmolecular and synthetic biology\ncomputational biology\ngenetics and genomics\npathology\nimmunology\nmicrobiology\nneuroscience\ndevelopmental biology\nethology and behavior\nzoology\nplant biology\nindustrial biotechnology\npharmacology\nengineering\nvirology\nmachine learning\nchemical biology\nnanomedicine\naging\necology and evolution\nvaccinology\nepidemiology\nclinical trials,\nb) write a 2-sentences summary, focusing on the key innovation presented in that abstract.\n\nfor example:\nmy input = The spontaneous deamination of cytosine is a major source of transitions from C•G to T•A base pairs, which account for half of known pathogenic point mutations in humans. The ability to efficiently convert targeted A•T base pairs to G•C could therefore advance the study and treatment of genetic diseases. The deamination of adenine yields inosine, which is treated as guanine by polymerases, but no enzymes are known to deaminate adenine in DNA. Here we describe adenine base editors (ABEs) that mediate the conversion of A•T to G•C in genomic DNA. We evolved a transfer RNA adenosine deaminase to operate on DNA when fused to a catalytically impaired CRISPR–Cas9 mutant. Extensive directed evolution and protein engineering resulted in seventh-generation ABEs that convert targeted A•T base pairs efficiently to G•C (approximately 50% efficiency in human cells) with high product purity (typically at least 99.9%) and low rates of indels (typically no more than 0.1%). ABEs introduce point mutations more efficiently and cleanly, and with less off-target genome modification, than a current Cas9 nuclease-based method, and can install disease-correcting or disease-suppressing mutations in human cells. Together with previous base editors, ABEs enable the direct, programmable introduction of all four transition mutations without double-stranded DNA cleavage.\n\nyour output =\na. genetics and genomics\nb. A new base-editor that converts A-T to G-C, based on an RNA adenosine deaminase fused to catalitically-impaired CRISPR-Cas9. Base editors can install therapeutic mutations in genomic DNA in human cells with no double-strand break.\nready to start?"]
for user_message, bot_message in zip(history_user, history_bot):
messag.append({"role": "user", "content": str(user_message)})
messag.append({"role": "system", "content": str(bot_message)})
messag.append({"role": "user", "content": str(prompt)})
response = openai.ChatCompletion.create(
# please use gtp3.5 although gpt4 is much better for $$
model="gpt-3.5-turbo",
messages=messag
)
result = ''
for choice in response.choices:
result += choice.message.content
history_bot.append(result)
history_user.append(str(prompt))
return result
</code>
<code>
print(generate_response("The power of human language and thought arises from systematic compositionality—the algebraic ability to understand and produce novel combinations from known components. Fodor and Pylyshyn1 famously argued that artificial neural networks lack this capacity and are therefore not viable models of the mind. Neural networks have advanced considerably in the years since, yet the systematicity challenge persists. Here we successfully address Fodor and Pylyshyn’s challenge by providing evidence that neural networks can achieve human-like systematicity when optimized for their compositional skills. To do so, we introduce the meta-learning for compositionality (MLC) approach for guiding training through a dynamic stream of compositional tasks. To compare humans and machines, we conducted human behavioural experiments using an instruction learning paradigm. After considering seven different models, we found that, in contrast to perfectly systematic but rigid probabilistic symbolic models, and perfectly flexible but unsystematic neural networks, only MLC achieves both the systematicity and flexibility needed for human-like generalization. MLC also advances the compositional skills of machine learning systems in several systematic generalization benchmarks. Our results show how a standard neural network architecture, optimized for its compositional skills, can mimic human systematic generalization in a head-to-head comparison."))
</code>
<code>
def get_embedding(text_to_embed):
# Embed a line of text
response = openai.Embedding.create(
model= "text-embedding-ada-002",
input=[text_to_embed]
)
# Extract the AI output embedding as a list of floats
embedding = response["data"][0]["embedding"]
return embedding
</code>
<code>
review_df = pd.read_csv('Amazon_Unlocked_Mobile.csv')
review_df = review_df[['Reviews']]
print("Data shape: {}".format(review_df.shape))
display(review_df.head())
</code>
<code>
review_df = review_df[0:37]
review_df["embedding"] = review_df["Reviews"].astype(str).apply(get_embedding)
# Make the index start from 0
#review_df.reset_index(drop=True)
#review_df.head(10)
print(review_df["Reviews"][36])
</code>
<code>
def mse(a, b) :
answer = 0
for i in range(len(a)) :
answer += (a[i] - b[i])**2
return answer/len(a)
</code>
<code>
def closest_vector(n) :
answer = 100000000
answer_index = n
for i in range(37) :
if i != n :
possible_answer = mse(review_df['embedding'][n], review_df['embedding'][i])
if possible_answer <= answer :
answer = possible_answer
answer_index = i
return answer_index
</code>
<code>
closest_vector(23)
</code>
<code>
print(review_df['Reviews'][23])
</code>
<code>
print(review_df['Reviews'][closest_vector(23)])
</code>
|
{
"filename": "lauzhacktest.ipynb",
"repository": "ihchaeryu/LauzHack23-RHR",
"query": "transformed_from_existing",
"size": 31023,
"sha": ""
}
|
# Traveling-Waves-nb.ipynb
Repository: Hallatscheklab/PAM
# Traveling waves
Now that we have a basic understanding of the stochastic dynamics of growing populations, we would like to embed the population in space. In the simplest case, we can imagine that each particle is merely diffusing along a line, without any active motion in a certain direction. Then, when we start with one initial particle and $R_0>1$, we will observe the expansion of the population both in density and range. Of particular interest is the shape and dynamics of the most advanced part of the population - the expanding population frontier. These fronts can serve as quantitative models of range expansions, epidemic outbreaks or evolutionary adaptation. Below we focus on the leading order behavior of the speed and shape of the fronts. Our treatment of number fluctuations is adhoc, but hopefully illuminating. Rigorous treatments of the asymptotic dynamics can be found in the literature{cite}`hallatschek2011noisy` Brunet Derrida, Igor, Kessler, Desai Fisher.
Once we allow the population to be distributed in space, we need a scalar field, the number density $c(x,t)$ at position $x$ and time $t$, to describe the state of the population, rather than a single number (the total population size). This makes the problem infinite dimensional and, thus, very challenging in general.
Still, progress can be made in special cases, for example, in the absence of non-linearities, in which case the bulk of the population grows without bound, or in deterministic limits. It turns out, however, that both non-linearities and number fluctuations matter for a realistic description of traveling waves.
We will first ignore fluctuations entirely, incorporate dispersal and see what such mean-field theories would predict.
## Incorporating dispersal
Given a well-mixed model $\partial_t N=s N$ for the net-growth of a population, the simplest way to include space is to say that the growth law applies locally and dispersal locally conserves particle numbers. Mathematically, this can be written as
$$
\partial_t c(x,t)=s(x,t) c(x,t) - \partial_x j(x,t) \;,
$$
where $c(x,t)$ is the number density at position $x$ and time $t$. The second term on the right hand site is the one-dimensional divergence of a local particle current $j(x,t)$, which represents the rate of chance of the number density of $x$ due to local dispersal. When the current decreases with $x$, densities must increase -- hence the minus sign.
In the simplest case, we assume that the current is proportional to the gradient of the density,
$$
j(x,t)=-D \partial_x c \;.
$$
Naturally the current goes from the high to low density regions -- hence the minus sign. This ansatz corresponds to diffusion and represents a coarse-grained model of a population where individuals carry out unbiased short-range dispersal.
Local dispersal seems like a strong assumption. More generally, we may introduce a jump kernal $K(\Delta x)$ representing the rate at which an individual jumps from $x$ to $x+\Delta x$. In that case, we may formulate the dynamics as follows
$$
\partial_t c(x,t)=s(x,t) c(x,t) + \int_{y}K(x-y) (c(y,t)-c(x,t) \;.
$$
It turns out, the diffusion approximation is an accurate description on long time and length scales if the kernel is sufficiently rapidly decaying with distance. However, what "sufficiently short-range" means depends on the reaction term. For the case of no reaction, $s=0$, one finds super-diffusive behavior, so called Levy flights, if $K(\Delta x)$ has a diverging variance. With a reaction term, non-diffusive behavior occurs even if the variance is finite but strongly depends on the presence of noise (see slides).
## Ignoring non-linearities
If we ignore the non-linear population control and assume the growth rate $s$ is strictly constant,
$$
\partial_t c=D\partial_x^2 c+ s c\;,
$$ (mean-branching-random-walk)
we can easily solve the ensuing reaction-diffusion equation. Namely, by substituting $c(c,t)=\psi(c,t)e^{st}$, we can get rid of the reaction term and obtain a simple diffusion equation,
$$
\partial_t \psi=D\partial_x^2 \psi \;,
$$
which we know how to solve. For example, if we assume localized initial conditions, we get a widening Gaussian with variance growing like $2D t$. In terms of the number density, this solution reads
$$
c(x,t)=e^{s t}e^{\frac{-x^2}{4Dt}} (4\pi Dt)^{-1/2}\;.
$$
Although the solution has a well-defined peak, its tails extend to infinity. In any real simulation of discrete particles, there will instead be a most advanced particle.
We can try to estimate the position of the most advanced particle in an adhoc way by asking: "At what position will the density drop below some threshold density $\epsilon$?" This should occur for $x>\ell(t)$ where $c(\ell(t),t)=\epsilon$, yielding $\ell(t)\sim v_F t$ where $v_F$ is the so-called Fisher-Kolmogorov velocity,
$$
v_F=2\sqrt{Ds}\;.
$$
## Including population control
By estimating the most advanced individual, we have just estimated an effect of the discreteness of the particles: There's always a leading edge. Our answer for the asymptotic velocity of that leading edge is is even the asymptotically correct result for a branching random walk, whose first moment equation is linear and exactly given by {eq}`mean-branching-random-walk`.
However, the linearized model has the unrealistic feature of an bulk population that grows without bound. To limit growth, we will now re-introduce non-linear logistic control, which yields the so-called Fisher-Kolmogorov equation,
$$
\partial_t c=D\partial_x^2 c+s c \left(1-\frac cK\right) \;.
$$ (FKPP-eq)
Under this model, the population develops, after some relaxation time, a steadily moving wave train. In the co-moving frame, the population profile becomes steady,
$$
0=D\partial_x^2 c+v \partial_x c + s c \left(1-\frac cK\right) \;.
$$
In the tip of the wave, where $c\ll K$, we can focus on the linearized version of this equation, which admits exponentially decaying solutions $c\sim e^{-k x}$ where $k$ satisfying $D k^2-v k+r=0$. This quadratic equation has a solution with positive $k$ only if the speed is larger or equal to the Fisher velocity $v\geq v_F=2\sqrt{D r}$. Which of these velocities is realized however?
This so-called velocity selection problem requires some thought in general, but has an easy answer for localized initial conditions. In that case, we already know that the FKPP equation without non-linearity, which maps to the mean branching random walk, has a front that advances at speed equal to $v_F$. Obviously, adding a negative term to the right side cannot *accelerate* the wave. Therefore, we can conclude that even the above non-linear waves travel, asymptotically, at the speed of the classical Fisher wave speed. Indeed, a more careful analysis shows that any initial condition with compact support leads to waves with $v=v_F$ and to an asymptotic density profile $c\sim e^{v x/2 D}$.
```{figure} ../images/FKPP-rolling-ball-analogy.jpg
---
height: 300px
name: rolling-balls
---
Rolling ball analogy for the steady state equation describing the FKPP wave profile
```
```{admonition} Different types of traveling waves
Waves are called pulled if the most advanced individuals have the largest growth rate. The ball analogy for those waves looks as in Fig. X a with a ball running down a hill onto a plane. Pushed and, more recently discovered, semi-pushed{cite}`birzu2018fluctuations` waves have reduced growth rates in the wave tip. The corresponding ball analogy is sketched in Fig. Xb, where a ball runs from one peak through a valley to a lower laying hill.
```
## Effects of discreteness
Our discussion of traveling waves ignored the fact that individuals are discrete. An exponentially decaying density can only make sense in an average sense when the density drops below one and that then implies the existence of fluctuations. It turns out these fluctuations crucial influence the behavior of waves.
One can show that the correct noisy version of the above dynamics requires to leading order on the right hand side of the FKPP equation a noise term of the form $ \eta(x,t)\sqrt{c}$ where the $\eta$ is uncorrelated white noise, $\eta(x,t)\eta(x',t')=\delta(x-x')\delta(t-t')$.
The FKPP equation {eq}`FKPP-eq` amounts to approximating $\langle c^2\rangle=\langle c\rangle^2$. It turns out that this mean-field approximation is problematic. The velocity is reduced by a singular correction that cannot be obtained through perturbation analysis. More serious are the consequences for models of adaptation, which do not even have a finite wave speed in the mean-field approximation, and for the genealogical structures generated by these waves.
A lot of work has been done to implement the consequences of discreteness, most of them being adhoc at some level. Below I present an adhoc recipe that has given correct answers to leading order for many cases.
This method requires us to first extend our understanding of branching processes to space and introduce the concept of gene surfing.
### Simple code to simulate FKPP waves
<code>
import numpy as np
import matplotlib.pyplot as plt
# Constants
D = 0.1 # Diffusion coefficient
a = 1.0 # Fisher-Kolmogorov parameter
L = 30.0 # Length of the domain
T = 20.0 # Simulation time
dx = 0.1 # Spatial step size
dt = 0.01 # Time step size
sigma0 = 1 # initial width
N0 = 1 # initial cummulative value
# Calculate the number of spatial and temporal steps
N = int(L / dx) + 1
M = int(T / dt) + 1
# Initialize the solution matrix
u = np.zeros((M, N))
# Set the initial condition
u[0, :] = N0/(sigma0 * np.sqrt(2 * np.pi)) * np.exp( - (np.linspace(0, L, N)-L/2)**2 / (2 * sigma0**2))
#u[0, :] = np.sin(np.pi * np.linspace(0, L, N) / L)
# Apply the finite difference method
for k in range(M - 1):
for i in range(1, N - 1):
u[k + 1, i] = u[k, i] + dt * (
D * (u[k, i - 1] - 2 * u[k, i] + u[k, i + 1]) / dx**2 +
a * u[k, i] * (1 - u[k, i])
)
# Plot the results
x = np.linspace(0, L, N)
t = np.linspace(0, T, M)
X, T = np.meshgrid(x, t)
fig = plt.figure()
ax = fig.add_subplot(111, projection='3d')
ax.plot_surface(X, T, u, cmap='viridis')
ax.set_xlabel('x')
ax.set_ylabel('t')
ax.set_zlabel('u')
plt.show()
</code>
The above code is modified version of what ChatGPT gave me when I asked "write a simple python code to simulate a traveling Fisher Kolmogorov wave":
This code uses a finite difference method to solve the Fisher-Kolmogorov equation with diffusion. It initializes a matrix u to store the solution at each time step. The initial condition is set as a sine wave, and then the finite difference method is applied iteratively to calculate the solution at each time step. Finally, the results are plotted using a 3D surface plot.
Please note that this code is a basic implementation and may not handle certain edge cases or optimizations. You can modify the parameters and experiment with different initial conditions to observe the behavior of the traveling Fisher-Kolmogorov wave.
## Gene Surfing
Taking discreteness into account is difficult in general. Forward in time, a branching process gives rise to a genealogical tree (show figure). Each time a birth event happens, a lineage splits into two, and both lineages have to be followed to describe the downstream dynamics fully probabilistically. That attempt is feasible if birth and death rates are constant but often impossible if they depend on the stochastic dynamics itself.
In these many-body problems of a branching tree, one can obtain a simpler single-body problem by taking a retrospective view: Sample one individual at the final time, i.e. a "tip" of the genealogical tree, and follow its lineage backward in time. A lineage traveling backward in time has the advantage that it neither splits nor ends, i.e. we only have to deal the degrees of freedom of just one particle. That can be useful if one knows the field of growth rates and used as a tool to enforce consistency of the dynamics.
To make this concrete, imagine sampling a particle of the a traveling wave and following it's lineage backward in time. To describe the ancestral process of said lineage by the probability density $G(\xi,\tau|x,t)$ that an individual presently, at time t and located at x, has descended from an ancestor that lived at $\xi$ at the earlier time $\tau$. In this context, it is natural to choose time as increasing towards the past, $\tau>t$, and to consider $(\xi,\tau)$ and $(x,t)$ as final and initial state of the ancestral trajectory, respectively. With this convention, the distribution G satisfies the initial condition $G(\xi,\tau|x,t)=\delta(\xi-x)$.
```{admonition} Notation
For space-time trajectories, we will use the convention that co-ordinates in greek letters $(\xi,\tau)$ refer to the starting point of the trajectory and those in roman letters $(x,t)$ refer to the end point.
```
Since a lineage cannot branch or end, all that it can do is to move around. If this movement is continuous and memory-less, we obtained a biased diffusion process. To describe this process, we develop a biased diffusion equation for the Greens function $G$.
Since $G$ is a pdf, it has to satisfy a continuity equation,
$$
\partial_\tau G=-\partial_\xi j\;,
$$
which simply ensures that the lineage is somewhere (conservation of probability). The probability current $j(\xi,\tau)$ is given by
$$
j=-D_*\partial_\xi G+ v_* G \;,
$$
where the first term represents unbiased diffusion and the second part a deterministic bias with velocity $v_*(\xi,\tau)$.
We have the mathematical form of the diffusion equation, but what's the diffusivity and what's the bias? A simple calculation shows that the diffusivity, $D_*=D$ is the same as forward in time but that the bias is number density dependent,
$$
v_*(\xi,\tau)=2 D \partial_\xi\ln[c(\xi,\tau)]
$$
In the co-moving frame, we expect the particle current to vanish
$$
0=j=-D\partial_\xi G(\xi)+\left[v+2 D \partial_\xi \ln(c)\right] G(\xi)
$$
which predicts that the steady state distribution $G(\xi)$ of the common ancestors is given by
$$
G\propto c^2 e^{v \xi/D}
$$ (pdf-ancestors)
where the pre-factor is fixed by the normalization condition, $\int_\xi G=1$.
Using the mean-field solution $c \propto c^{-v \xi/(2D)}$ of the FKPP, one finds that $G$ should vanish everywhere because it is not normalizable.
```{figure} ../images/Distribution-of-common-ancestors.jpg
---
height: 300px
name: Doca
---
Measured bell-shaped distribution $G_\infty(\xi)$ as function of position $\xi$ relative to the FKPP wave front (sigmoidal lines) for varying carrying capacities $N$.
```
But simulations of stochastic traveling waves show that this steady state distribution is a bell-like shape in the wave tip, see {numref}`Doca`. Another hint that the mean-field solution to the number density is incorrect, in fact completely off.
## Branching Random Walks
It turns out that a more appropriate approximation to noisy traveling waves can be obtained from taking a particle viewpoint from the beginning.
One of the most extreme consequences of discreteness is that lineages can go extinct. In fact, when birth and death rates are nearly the same, the probability $u$ of survival is very low, of order the relative growth rate difference $s$. We arrived at this conclusion by deriving and solving a differential equation for the survival probability, which read
$$
-\partial_\tau u(t|\tau)=s u(t|\tau)- u^2(t|\tau) \;.
$$
Generalizing this equation to the case of diffusion and spatially varying growth rates $s(\xi,\tau)$, one obtains
$$
-\partial_\tau u (x,t|\xi,\tau)=D\partial_\xi u^2+s(\xi,\tau) u- u^2 \;.
$$ (u-FKPP)
Deriving this equation is a useful exercise, for which one has to retrace the same steps as for the non-spatial equation with the addition that one needs to account for the rates of dispersal. Notice that {eq}`u-FKPP` has a similar shape as the FKPP equation.
In the frame co-moving with the wave traveling in the $-x$ direction backward in time, we have
$$
0 =D\partial_\xi u+ v \partial_\xi u+s(\xi) u- u^2 \;,
$$ (u-FKPP)
where $s(\xi)=1-c(\xi)$ is given in terms of the density. To close this equation, we need an expression for the number density $c$ in terms of the survival probability $u$. Such a connection is provided by equation {eq}`pdf-ancestors` for the distribution of common ancestors, once we recognize that
$$
G(x,\infty|\xi,\tau)=u(\xi,\tau) c(\xi,\tau) \;.
$$ (G-u-c)
To see {eq}`G-u-c`, notice that the probability that, at time $\tau$, the common ancestor of future generations is at $\xi$ is the same as the probability that any particle at $\xi$ is going to survive forever, which is given by $u c$.
Combining Eq.s {eq}`u-FKPP`, {eq}`pdf-ancestors` and {eq}`G-u-c`, we obtain
$$
0 =D\partial_x c^2+ v \partial_x c+r c(1-c)- \frac{c^2 e^{v x/D}}{\int_x c^2 e^{v x/D}}\;.
$$ (c-cutoff)
The third term on the right-hand side furnishes the major difference to the naive mean-field approach. One might think, at first sight, that this non-linear term can be ignored in the tip of the wave, where the density decays to 0 and $c^2\ll c$. But the exponential amplification factor ensures that this factor becomes a leading order term sufficiently far out in the tip of the wave.
## Consequences of tip fluctuations
### FKPP waves
Assuming an overall decay $c \sim N e^{-v x/(2 D)}$, it is easy to see that the quadratic cut-off term becomes of comparable to the first order terms when $x_c \sim 2\ln(N)D/v$. For $x>x_c$, the density decays faster, $c\sim e^{-v x/D}$, just as if there is no growth beyond $x_c$. I.e. the leading order effect of the fluctuation-induced term is to introduce a growth cutoff at $x_c$. Therefore, the linearized equation
$$
\partial_t c =D\partial_x^2 c+ v \partial_x c+r c
$$ (FKPP-linear)
has to be solved between $x=0$ and $x=x_c$. To do this, it is convenient to introduce the variable transform
$$
c(x,t)=\phi(x,t) e^{-\frac{vx}{2D}}
$$ (var-trafo)
through which {eq}`FKPP-linear` is transformed into
$$
\partial_t \phi =D\partial_x^2 \phi+ \left(r-\frac{v^2}{4D} \right) \phi\;,
$$ (FKPP-linear-trafo)
which has a *Hermitian* operator on the RHS. {eq}`FKPP-linear-trafo` has the shape of a Schroedinger equation in imaginary time. The non-linearities of the original equation {eq}`c-cutoff` effectively generate two absorbing boundary conditions for $\phi(x)$, one at $x=O(\ln N)$ and one at $x=O(1)$.
We can solve equation {eq}`FKPP-linear-trafo` subject to these boundary conditions in terms of normal modes. To ensure a steady state in the co-moving frame, we have to demand that the ground state (lowest mode) has vanishing energy. All higher modes have higher energy, and therefore decay over time. The higher the mode number (the more nodes the eigen function has / the more rapidly it varies in space) the faster its relaxation to 0. Since the growth rate is constant between the two absorbing boundaries, we essentially have the mode spectrum of a particle in a 1D-box, which are cosine modes.
To fix the wave speed, we focus on the ground state,
$$
\phi_0(x)\sim \sin\left(\frac{\pi x}{x_c}\right)
$$
which solves {eq}`FKPP-linear-trafo` at steady state ($\partial_t \phi=0$) if
$$
0=-D\left(\frac{\pi }{x_c}\right)^2 + \left(r-\frac{v^2}{4D}\right)
$$
implying that
$$
v = 2\sqrt{D r}\left(1-\frac{\pi^2 D}{x_c^2 r}\right) = 2\sqrt{D r}\left(1-\frac{\pi^2 v^2 }{4\ln(N)^2 D r}\right) \;.
$$
Solving this equation, we obtain to leading order
$$
v = v_F\left(1-\frac{\pi^2 }{\ln(N)^2 }\right) \;.
$$
So, the cutoff leads to a reduction of the wave speed, which only slowly vanishes as $\ln N$ goes to infinity. In particular, it cannot be captured by a perturbation theory in the noise strength $N^{-1}$.
The consequences of the cutoff on genealogical processes are even more pronounced. Without going into the details, we merely argue intuitively here. Due to the the second cutoff, lineages are localized on a scale $\ln N$ and caused to coalesce on a time scale $T_c\sim \ln^3 N$, which diverges when $N\to \infty$.
## Fitness waves
Traveling waves not only occur in real space. They are often used to describe how the distribution of a trait, such as growth rate, height, strength etc. across a population shifts over time in response to a changing environment or in response to self-organization. The most classical of these waves is a wave of adaptation, where growth rate, or fitness, shifts towards larger values in response to the occurrence of fitter types (due to mutations) and their take over within the population.
A simple model to describe this situation is
$$
\partial_t c_n = U \left(c_{n-1}-c_{n}\right)+ s(n-\overline{n})c + \eta \sqrt{c_n} \;,
$$ (eq-staircase)
where it is assumed that beneficial mutations all beneficial mutations have the same fitness effect $s$ and occur at rate $U$. The overline represents the population mean, $\overline{f_n}\equiv \sum_n c_n f_n$, so that $s\overline{n}$ is the mean fitness.
Two simple arguments constrain the leading order speed of these waves.
#### Adaptation speed depends on fitness variance
Multiplying {eq}`eq-staircase` with $s(n-\overline{n})$, integrating over $n$ and averaging over the noise yields
$$
s \partial_t\overline n(t) = s^2\overline{ (n-\overline{n})^2} \;,
$$
i.e. the rate of increase in fitness (LHS) equals the variance in fitness (RHS), or
$$
v=\sigma^2\;.
$$
When R. A. Fisher stumbled over this simple observation, he was apparently so excited that he called it "the fundamental law of natural selection", although it was soon shown that this equation is neither fundamental nor a law.
#### The tip of the wave
The density profile in the co-moving frame looks, to a good approximation like a Gaussian. But, because of the discreteness of individuals, this Gaussian has to be cut-off at the fitness value $q$ of the most fit individuals. This tip of the wave can be estimated from $e^{-(q s)^2 /(2 \sigma^2)}N\sim 1$ giving us
$$
q\sim s^{-1}\sqrt{2\sigma^2 \ln N} \;.
$$
At the wave tip, we can estimate how long it takes for the next bin to be established, by
$$
(q+1) s U\int^T dt' \frac{e^{q s t'}}{q s } \sim 1 \;,
$$
giving us
$$
T=\frac{\log{U^{-1}}}{qs}=\frac{\log{U^{-1}}}{\sqrt{2\sigma^2 \ln N}}\;.
$$
#### Self-consistency
The wave speed $v\approx \sigma^2$ to be consistent with the weighting time $T$, we have to demand $s/v\sim T$, implying
$$
v\sim 2 s^2 \frac{\ln N }{\ln^2{U}}
$$
Note that, for these types of waves, number fluctuations not only cause a correction to the wave speed. Without taking fluctuations into account ($N\to\infty$), the wave speed diverges, as it turns out, in finite time.
#### Naive expectation
Yet, the wave speed increases very slowly with population size, namely just logarithmically. According to classical population genetics, one would expect a linear increase, $v= U N s$, based on the following computation. New beneficial mutations arise at a rate $U N$, and each one of those mutations has a survival probability of $s$. What's wrong with that computation? Respectively, when is this computation correct?
<code>
import numpy as np
import matplotlib.pyplot as plt
# Constants
D = 0.1 # Diffusion coefficient
a = 1.0 # Fisher-Kolmogorov parameter
L = 30.0 # Length of the domain
T = 20.0 # Simulation time
dx = 0.1 # Spatial step size
dt = 0.01 # Time step size
noisestrength = .001
sigma0 = 1 # initial width
# Calculate the number of spatial and temporal steps
N = int(L / dx) + 1
M = int(T / dt) + 1
# Initialize the solution matrix
u = np.zeros((M, N))
# Set the initial condition
#u[0, :] = N0/(sigma0 * np.sqrt(2 * np.pi)) * np.exp( - (np.linspace(0, L, N)-L/2)**2 / (2 * sigma0**2))
u[0, :] = np.exp( - (np.linspace(0, L, N)-L/2)**2 / (2 * sigma0**2))
#u[0, :] = np.sin(np.pi * np.linspace(0, L, N) / L)
# Apply the finite difference method
for k in range(M - 1):
for i in range(1, N - 1):
u[k + 1, i] = np.random.poisson((u[k, i] + dt * (
D * (u[k, i - 1] - 2 * u[k, i] + u[k, i + 1]) / dx**2 +
a * u[k, i] * (1 - u[k, i]))) / noisestrength) * noisestrength
# Plot the results
x = np.linspace(0, L, N)
t = np.linspace(0, T, M)
X, T = np.meshgrid(x, t)
fig = plt.figure()
ax = fig.add_subplot(111, projection='3d')
ax.plot_surface(X, T, u, cmap='viridis')
ax.set_xlabel('x')
ax.set_ylabel('t')
ax.set_zlabel('u')
plt.show()
</code>
```{admonition} Speed of adaptation
The speed of adaptation scales with the log of the population size, much slower than naively expected (based on sequential selection) yet still diverging if number fluctuations are neglected ($N\to \infty$).
```
## Comment
Why do we prefer equation XXX with the emerging cutoff over equation YYY? Both treatments are approximate and uncontrolled by a small parameter.
The primary reason is the nature of the assumptions being made. The naive mean field theory assumes that growth rates are given by $r(1-\langle c\rangle)$. By contrast, the gene-surfing-based theory is based on knowing the mean of the log, $\langle v_* \rangle = 2 D \partial_x \langle \ln c\rangle$. The log of a quantity usually fluctuates much less than the quantity itself. The flaw of this intuitive argument is, however, there are realizations where the density drops to 0 at any x, which should dominate the mean of the log. To the rescue comes the fact that we need the quantity $\partial_x \ln c(x)$ only if a lineage is actually present at $x$, so that it can be sampled. Thus, we if we interpret the average $\langle \ln c(x)\rangle$ as conditional on a lineage being present at $x$, a divergence is prevented.
Well, if such hand waving still leaves you uneasy, there is a way to formulate non-linear noisy waves with a closed first moment{cite}`hallatschek2011noisy`, which gives rise to the exact same cutoff as in Eq. XXX except for a factor of $2$! This method can be generalized to obtain models that, for any integer $n$, have a closed set of $n$ moment equations{cite}`hallatschek2016collective`.
## Problems
1. Estimate the speed of a FKPP wave traveling down a narrow corridor of width $W$. Assume the walls of the corridor to be absorbing.
2. Use the analogy of traveling waves to imaginary time Schroedinger equation to make predictions about traveling waves that involve standard solutions of quantum mechanics problems you know (using e.g. tunneling, defects, etc.).
3. Simulate an FKPP or fitness wave with finite particle numbers and measure speed and wave diffusion as function of $\ln N$
4. Extend the theory of branching random walks to offspring distributions with diverging moments.
## Citations
```{bibliography}
```
|
{
"filename": "Traveling-Waves-nb.ipynb",
"repository": "Hallatscheklab/PAM",
"query": "transformed_from_existing",
"size": 347696,
"sha": ""
}
|
# Career.ipynb
Repository: annanya-mathur/Career-Prediction
<code>
!pip install pandas
!pip install sklearn
!pip install xlrd
from sklearn.model_selection import train_test_split
from sklearn.linear_model import LinearRegression
import numpy as np
import pandas as pd
from sklearn.ensemble import RandomForestRegressor
</code>
<code>
df=pd.read_csv('exams (2).csv')
df.head(5)
</code>
<code>
career=pd.read_csv('Career_Choices.csv')
career.head(15)
</code>
<code>
df.shape
</code>
<code>
career.shape
</code>
<code>
X=df[['aptitude score','test preparation course']]
</code>
<code>
Y = df[['GradePoints']]
</code>
<code>
X_train,X_test,Y_train,Y_test =train_test_split(X,Y,test_size=0.3)
</code>
<code>
X_train
</code>
<code>
Y_test
</code>
<code>
rf=rf=RandomForestRegressor()
</code>
<code>
X_train.info()
</code>
<code>
Y_train.info()
</code>
<code>
rf.fit(np.array(X_train).reshape(-1,2),np.array(Y_train).reshape(-1,1))
</code>
<code>
a=int(input("Enter Aptitude score of student "))
b=int(input("Say whether student completed test preparation : 1 for Yes & 0 for No "))
r=np.array(rf.predict(np.array([[a,b]])))
</code>
<code>
print (r)
</code>
<code>
if r>=0.9 and r<=1.0 :
choices = career[['90% - 100%','70% - 90%','50% - 70%','40% - 50%','10% - 40%']]
elif r >=0.7 and r<=0.9:
choices = career[['70% - 90%','50% - 70%','40% - 50%','10% - 40%']]
elif r>= 0.5 and r<=0.7:
choices = career[['50% - 70%','40% - 50%','10% - 40%']]
elif r>=0.4 and r<=0.5:
choices = career[['40% - 50%','10% - 40%']]
else :
choices =career[['10% - 40%']]
final=choices.values.tolist()
</code>
<code>
type(choices)
</code>
<code>
n=len(final)
n
</code>
<code>
for i in final:
for h in i:
print(h)
</code>
<code>
import pickle
with open("career2.pkl","wb") as f:
pickle.dump(rf,f)
loader_model=pickle.load(open("career2.pkl","rb"))
result=loader_model.score(X_test,Y_test)
print(result)
</code>
<code>
h=pd.read_pickle('career2.pkl')
</code>
<code>
h
</code>
|
{
"filename": "Career.ipynb",
"repository": "annanya-mathur/Career-Prediction",
"query": "transformed_from_existing",
"size": 36414,
"sha": ""
}
|
# project_drug.ipynb
Repository: satish2705/major
<code>
import pandas as pd
import numpy as np
import random
# Generate synthetic dataset
num_samples = 1000
# Patient Information
patient_ids = [f"P{str(i).zfill(5)}" for i in range(1, num_samples + 1)]
ages = np.random.randint(18, 90, num_samples)
genders = np.random.choice(["Male", "Female"], num_samples)
medical_history = np.random.choice(["Diabetes", "Hypertension", "Cancer", "None"], num_samples)
drug_names = np.random.choice(["DrugA", "DrugB", "DrugC", "DrugD"], num_samples)
dosages = np.random.randint(50, 500, num_samples)
treatment_durations = np.random.randint(5, 60, num_samples)
effectiveness = np.random.uniform(0, 100, num_samples)
side_effects = np.random.choice(["None", "Nausea", "Dizziness", "Fatigue"], num_samples)
disease_types = np.random.choice(["Lung Cancer", "Breast Cancer", "Diabetes", "Heart Disease"], num_samples)
genetic_markers = np.random.choice(["MarkerA", "MarkerB", "MarkerC", "MarkerD"], num_samples)
# Treatment Outcome
response_to_treatment = np.random.choice(["Positive", "Negative"], num_samples)
success_rates = np.random.uniform(50, 100, num_samples)
# Create DataFrame
dataset = pd.DataFrame({
"Patient_ID": patient_ids,
"Age": ages,
"Gender": genders,
"Medical_History": medical_history,
"Drug_Name": drug_names,
"Dosage_mg": dosages,
"Treatment_Duration_days": treatment_durations,
"Effectiveness_%": effectiveness,
"Side_Effects": side_effects,
"Disease_Type": disease_types,
"Genetic_Marker": genetic_markers,
"Response_to_Treatment": response_to_treatment,
"Success_Rate_%": success_rates
})
# Save to CSV
dataset.to_csv("synthetic_medical_dataset.csv", index=False)
print("Dataset generated successfully!")
</code>
<code>
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras.models import Sequential
from tensorflow.keras.layers import Conv1D, MaxPooling1D, Flatten, Dense, Dropout, BatchNormalization
import pandas as pd
import numpy as np
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import LabelEncoder, StandardScaler
# Load dataset
dataset = pd.read_csv("synthetic_medical_dataset.csv")
# Encode categorical variables
label_encoders = {}
categorical_columns = ["Gender", "Medical_History", "Drug_Name", "Side_Effects", "Disease_Type", "Genetic_Marker", "Response_to_Treatment"]
for col in categorical_columns:
le = LabelEncoder()
dataset[col] = le.fit_transform(dataset[col])
label_encoders[col] = le
# Selecting features and target variables
X = dataset.drop(columns=["Patient_ID", "Response_to_Treatment"]).values
y = dataset["Response_to_Treatment"].values
# Normalize features
scaler = StandardScaler()
X = scaler.fit_transform(X)
# Reshape for CNN input (assuming 1D features per patient)
X = np.expand_dims(X, axis=2)
# Split dataset
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Build CNN model
model = Sequential([
Conv1D(filters=64, kernel_size=3, activation='relu', input_shape=(X.shape[1], 1)),
BatchNormalization(),
MaxPooling1D(pool_size=2),
Dropout(0.2),
Conv1D(filters=128, kernel_size=3, activation='relu'),
BatchNormalization(),
MaxPooling1D(pool_size=2),
Dropout(0.3),
Flatten(),
Dense(128, activation='relu'),
Dropout(0.4),
Dense(1, activation='sigmoid') # Binary classification
])
# Compile the model
model.compile(optimizer='adam', loss='binary_crossentropy', metrics=['accuracy'])
# Train the model
model.fit(X_train, y_train, epochs=30, batch_size=32, validation_data=(X_test, y_test))
# Evaluate the model
test_loss, test_acc = model.evaluate(X_test, y_test)
print(f"Test Accuracy: {test_acc:.2f}")
# Save the model
model.save("cnn_drug_discovery_model.h5")
print("Model saved successfully!")
</code>
<code>
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras.models import Sequential, load_model
from tensorflow.keras.layers import Conv1D, MaxPooling1D, Flatten, Dense, Dropout, BatchNormalization
import numpy as np
from sklearn.preprocessing import StandardScaler, LabelEncoder
# Load the trained model
model_path = r"cnn_drug_discovery_model.h5"
model = load_model(model_path)
print("Model loaded successfully!")
# Define label encoders for categorical values
label_encoders = {
"Gender": LabelEncoder().fit(["Male", "Female"]),
"Medical_History": LabelEncoder().fit(["Diabetes", "Hypertension", "Cancer", "None"]),
"Drug_Name": LabelEncoder().fit(["DrugA", "DrugB", "DrugC", "DrugD"]),
"Side_Effects": LabelEncoder().fit(["None", "Nausea", "Dizziness", "Fatigue"]),
"Disease_Type": LabelEncoder().fit(["Lung Cancer", "Breast Cancer", "Diabetes", "Heart Disease"]),
"Genetic_Marker": LabelEncoder().fit(["MarkerA", "MarkerB", "MarkerC", "MarkerD"])
}
# Define a scaler (use values from training phase if available)
scaler = StandardScaler()
# Example input values
input_data = {
"Age": 45,
"Gender": "Male",
"Medical_History": "Diabetes",
"Drug_Name": "DrugA",
"Dosage_mg": 200,
"Treatment_Duration_days": 30,
"Effectiveness_%": 85.4,
"Side_Effects": "Nausea",
"Disease_Type": "Lung Cancer",
"Genetic_Marker": "MarkerB"
}
# Encode categorical values
for key in label_encoders:
if key in input_data:
input_data[key] = label_encoders[key].transform([input_data[key]])[0]
# Convert input data to array
input_array = np.array(list(input_data.values())).reshape(1, -1)
# Normalize input features (use values from training phase if available)
input_array = scaler.fit_transform(input_array) # Use transform() instead of fit_transform() if scaler was previously trained
# Reshape for CNN input
input_array = np.expand_dims(input_array, axis=2)
# Make prediction
prediction = model.predict(input_array)
predicted_class = (prediction > 0.5).astype(int)
print(f"Predicted Response: {predicted_class[0][0]}")
</code>
<code>
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras.models import Sequential, load_model
from tensorflow.keras.layers import Conv1D, MaxPooling1D, Flatten, Dense, Dropout, BatchNormalization
import numpy as np
from sklearn.preprocessing import StandardScaler, LabelEncoder
# Load the trained model
model_path = r"cnn_drug_discovery_model.h5"
model = load_model(model_path)
print("Model loaded successfully!")
# Define label encoders for categorical values
label_encoders = {
"Gender": LabelEncoder().fit(["Male", "Female"]),
"Medical_History": LabelEncoder().fit(["Diabetes", "Hypertension", "Cancer", "None"]),
"Drug_Name": LabelEncoder().fit(["DrugA", "DrugB", "DrugC", "DrugD"]),
"Side_Effects": LabelEncoder().fit(["None", "Nausea", "Dizziness", "Fatigue"]),
"Disease_Type": LabelEncoder().fit(["Lung Cancer", "Breast Cancer", "Diabetes", "Heart Disease"]),
"Genetic_Marker": LabelEncoder().fit(["MarkerA", "MarkerB", "MarkerC", "MarkerD"])
}
# Define a scaler (use values from training phase if available)
scaler = StandardScaler()
# Example input values for drug effectiveness prediction
input_data = {
"Age": 45,
"Gender": "Male",
"Medical_History": "Diabetes",
"Drug_Name": "DrugB",
"Dosage_mg": 100,
"Treatment_Duration_days": 40,
"Effectiveness_%": 15.4,
"Side_Effects": "Nausea",
"Disease_Type": "Lung Cancer",
"Genetic_Marker": "MarkerB"
}
# Encode categorical values
for key in label_encoders:
if key in input_data:
input_data[key] = label_encoders[key].transform([input_data[key]])[0]
# Convert input data to array
input_array = np.array(list(input_data.values())).reshape(1, -1)
# Normalize input features (use values from training phase if available)
input_array = scaler.fit_transform(input_array) # Use transform() instead of fit_transform() if scaler was previously trained
# Reshape for CNN input
input_array = np.expand_dims(input_array, axis=2)
# Make prediction for drug effectiveness
prediction = model.predict(input_array)
effectiveness_score = prediction[0][0] * 100 # Convert to percentage
print(f"Predicted Drug Effectiveness: {effectiveness_score:.2f}%")
</code>
<code>
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras.models import Sequential
from tensorflow.keras.layers import Conv1D, MaxPooling1D, Flatten, Dense, Dropout, BatchNormalization
import pandas as pd
import numpy as np
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import LabelEncoder, StandardScaler
import joblib # Import joblib for saving the scaler
# Load dataset
dataset = pd.read_csv("synthetic_medical_dataset.csv")
# Encode categorical variables
label_encoders = {}
categorical_columns = ["Gender", "Medical_History", "Drug_Name", "Side_Effects", "Disease_Type", "Genetic_Marker", "Response_to_Treatment"]
for col in categorical_columns:
le = LabelEncoder()
dataset[col] = le.fit_transform(dataset[col])
label_encoders[col] = le
# Selecting features and target variables
# Selecting features and target variables
import joblib
# Selecting features (Ensure "Response_to_Treatment" is excluded)
X = dataset.drop(columns=["Patient_ID", "Response_to_Treatment"]).values
y = dataset["Response_to_Treatment"].values
# Save feature names (to ensure consistency during prediction)
feature_names = list(dataset.drop(columns=["Patient_ID", "Response_to_Treatment"]).columns)
joblib.dump(feature_names, "feature_names.pkl") # Save feature names
# Normalize features
scaler = StandardScaler()
X = scaler.fit_transform(X)
# Save the trained scaler
joblib.dump(scaler, "scalers.pkl")
print("Scaler and feature names saved successfully!")
# Reshape for CNN input (assuming 1D features per patient)
X = np.expand_dims(X, axis=2)
# Split dataset
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Build CNN model
model = Sequential([
Conv1D(filters=64, kernel_size=3, activation='relu', input_shape=(X.shape[1], 1)),
BatchNormalization(),
MaxPooling1D(pool_size=2),
Dropout(0.2),
Conv1D(filters=128, kernel_size=3, activation='relu'),
BatchNormalization(),
MaxPooling1D(pool_size=2),
Dropout(0.3),
Flatten(),
Dense(128, activation='relu'),
Dropout(0.4),
Dense(1, activation='sigmoid') # Binary classification
])
# Compile the model
model.compile(optimizer='adam', loss='binary_crossentropy', metrics=['accuracy'])
# Train the model
model.fit(X_train, y_train, epochs=30, batch_size=32, validation_data=(X_test, y_test))
# Evaluate the model
test_loss, test_acc = model.evaluate(X_test, y_test)
print(f"Test Accuracy: {test_acc:.2f}")
# Save the model
model.save("cnn_drug_discovery_model.h5")
print("Model saved successfully!")
</code>
<code>
import joblib
import numpy as np
import tensorflow as tf
from tensorflow.keras.models import load_model
from sklearn.preprocessing import LabelEncoder
# Load model, scaler, and feature names
model = load_model("cnn_drug_discovery_model.h5")
scaler = joblib.load("scalers.pkl")
expected_features = joblib.load("feature_names.pkl") # Load expected feature names
print("Model, scaler, and feature names loaded successfully!")
# Print expected features for debugging
print("Expected Features from Training:", expected_features)
# Define label encoders
label_encoders = {
"Gender": LabelEncoder().fit(["Male", "Female"]),
"Medical_History": LabelEncoder().fit(["Diabetes", "Hypertension", "Cancer", "None"]),
"Drug_Name": LabelEncoder().fit(["DrugA", "DrugB", "DrugC", "DrugD"]),
"Side_Effects": LabelEncoder().fit(["None", "Nausea", "Dizziness", "Fatigue"]),
"Disease_Type": LabelEncoder().fit(["Lung Cancer", "Breast Cancer", "Diabetes", "Heart Disease"]),
"Genetic_Marker": LabelEncoder().fit(["MarkerA", "MarkerB", "MarkerC", "MarkerD"])
}
# Example input values (Ensure all expected features are included)
input_data = {
"Age": 45,
"Gender": "Male",
"Medical_History": "Diabetes",
"Drug_Name": "DrugA",
"Dosage_mg": 900,
"Treatment_Duration_days": 10,
"Effectiveness_%": 95.4,
"Side_Effects": "Nausea",
"Disease_Type": "Lung Cancer",
"Genetic_Marker": "MarkerB"
}
# Encode categorical values
for key in label_encoders:
if key in input_data:
input_data[key] = label_encoders[key].transform([input_data[key]])[0]
# Ensure input_data has all expected features
for feature in expected_features:
if feature not in input_data:
print(f"Warning: Missing feature '{feature}' in input data. Assigning default value 0.")
input_data[feature] = 0 # Default value (adjust if needed)
# Convert input data to NumPy array in correct order
input_array = np.array([input_data[feature] for feature in expected_features]).reshape(1, -1)
# Verify feature count consistency
if input_array.shape[1] != scaler.n_features_in_:
raise ValueError(f"Feature mismatch: Expected {scaler.n_features_in_}, but got {input_array.shape[1]}.")
# Normalize input using the pre-trained scaler
input_array = scaler.transform(input_array)
# Reshape for CNN input
input_array = np.expand_dims(input_array, axis=2)
# Make prediction
prediction = model.predict(input_array)
effectiveness_score = prediction[0][0] * 100 # Convert to percentage
print(f"Predicted Drug Effectiveness: {effectiveness_score:.2f}%")
</code>
|
{
"filename": "project_drug.ipynb",
"repository": "satish2705/major",
"query": "transformed_from_existing",
"size": 46092,
"sha": ""
}
|
# ml_basics_1.ipynb
Repository: timeowilliams/Responsible-ai
<code>
pip install numpy scikit-learn
</code>
<code>
import numpy as np
from sklearn.linear_model import LogisticRegression
from sklearn.feature_extraction.text import CountVectorizer
# Sample training data
papers = [
"COVID vaccine clinical trials", # Medical
"vaccine efficacy study results", # Medical
"psychological impact of lockdown", # Psychology
"anxiety depression treatment study", # Psychology
"vaccine side effects analysis", # Medical
"mental health during pandemic", # Psychology
"antibody response in patients", # Medical
"cognitive behavioral therapy research" # Psychology
]
# Labels: 1 for Medical, 0 for Psychology
labels = np.array([1, 1, 0, 0, 1, 0, 1, 0])
# Convert text to numbers using simple word counting
vectorizer = CountVectorizer()
X = vectorizer.fit_transform(papers)
# Train the model
model = LogisticRegression()
model.fit(X, labels)
# Function to predict journal and show reasoning
def predict_paper(paper_text):
# Convert paper text to numbers
paper_features = vectorizer.transform([paper_text])
# Get prediction and probability
prediction = model.predict(paper_features)[0]
probabilities = model.predict_proba(paper_features)[0]
# Get important words and their weights
feature_names = vectorizer.get_feature_names_out()
weights = model.coef_[0]
word_weights = dict(zip(feature_names, weights))
# Print results
print(f"\nPaper text: '{paper_text}'")
print(f"Predicted journal: {'Medical' if prediction == 1 else 'Psychology'}")
print(f"Confidence: Medical={probabilities[1]:.2%}, Psychology={probabilities[0]:.2%}")
# Show top words that influenced the decision
print("\nTop words influencing the decision:")
for word in paper_text.split():
if word in word_weights:
print(f"- '{word}': weight={word_weights[word]:.2f}")
# Test some examples
test_papers = [
"new vaccine development study",
"depression and anxiety research",
"COVID treatment clinical trial"
]
for paper in test_papers:
predict_paper(paper)
</code>
<code>
import numpy as np
from sklearn.feature_extraction.text import CountVectorizer
class SimpleJournalClassifier:
def __init__(self, learning_rate=0.1):
self.learning_rate = learning_rate
self.vectorizer = CountVectorizer()
self.weights = None
self.bias = 0
def train_one_example(self, paper, true_label, print_details=True):
# Convert paper text to word counts
features = self.vectorizer.transform([paper]).toarray()[0]
print(f"\nFeatures for paper '{paper}':" + "\n" + str(features))
# Make prediction with current weights
prediction = np.dot(features, self.weights) + self.bias
predicted_label = 1 if prediction > 0 else 0
if print_details:
print(f"\nPaper: '{paper}'")
print(f"True label: {'Medical' if true_label == 1 else 'Psychology'}")
print(f"Predicted: {'Medical' if predicted_label == 1 else 'Psychology'}")
# Show current weights for words in this paper
print("\nCurrent word weights:")
words = paper.split()
for word in words:
if word in self.vectorizer.vocabulary_:
idx = self.vectorizer.vocabulary_[word]
print(f"- '{word}': {self.weights[idx]:.2f}")
# If prediction is wrong, adjust weights
if predicted_label != true_label:
error = true_label - predicted_label
# Update weights for each word
self.weights += self.learning_rate * error * features
self.bias += self.learning_rate * error
if print_details:
print("\nPrediction was wrong! Adjusting weights...")
print("New word weights:")
for word in words:
if word in self.vectorizer.vocabulary_:
idx = self.vectorizer.vocabulary_[word]
print(f"- '{word}': {self.weights[idx]:.2f}")
return predicted_label == true_label
def train(self, papers, labels, epochs=3):
# First, build vocabulary and convert to number format
self.vectorizer.fit(papers)
# Initialize weights to small random values
vocab_size = len(self.vectorizer.vocabulary_)
self.weights = np.random.randn(vocab_size) * 0.01
print("Initial random weights:")
print(dict(zip(self.vectorizer.get_feature_names_out(), self.weights.round(2))))
# Training loop
for epoch in range(epochs):
print(f"\nEpoch {epoch + 1}:")
correct = 0
for paper, label in zip(papers, labels):
if self.train_one_example(paper, label):
correct += 1
print(f"\nAccuracy this epoch: {correct/len(papers):.0%}")
# Training data
papers = [
"vaccine clinical trials", # Medical
"depression therapy study", # Psychology
"vaccine patient response", # Medical
"anxiety treatment research" # Psychology
]
labels = [1, 0, 1, 0] # 1 for Medical, 0 for Psychology
# Create and train classifier
classifier = SimpleJournalClassifier(learning_rate=0.1)
classifier.train(papers, labels)
# Test the final model
print("\nTesting final model:")
test_papers = [
"new vaccine study",
"depression analysis"
]
for paper in test_papers:
features = classifier.vectorizer.transform([paper]).toarray()[0]
prediction = np.dot(features, classifier.weights) + classifier.bias
print(f"\nPaper: '{paper}'")
print(f"Prediction: {'Medical' if prediction > 0 else 'Psychology'}")
# Show weights of words in test paper
print("Word weights that influenced this decision:")
for word in paper.split():
if word in classifier.vectorizer.vocabulary_:
idx = classifier.vectorizer.vocabulary_[word]
print(f"- '{word}': {classifier.weights[idx]:.2f}")
</code>
|
{
"filename": "ml_basics_1.ipynb",
"repository": "timeowilliams/Responsible-ai",
"query": "transformed_from_existing",
"size": 19006,
"sha": ""
}
|
# 00_Setup_1.ipynb
Repository: tebe-nigrelli/MMN-Group-Project
This file sets up the various variables and functions that are used at
every point in the project.
The contents of this file are also made available via `import dataset`
from `dataset.py`.
# Imports
Various useful builtins:
<code>
from typing import *
import functools
import pathlib
</code>
External packages:
<code>
import pandas as pd
import numpy as np
</code>
Our main workhorse, `allensdk`:
<code>
import allensdk
from allensdk.brain_observatory.ecephys.ecephys_project_cache import EcephysProjectCache
from allensdk.brain_observatory.ecephys.ecephys_session import EcephysSession
</code>
Local modules:
<code>
from constraints import *
</code>
# Global Variables
We have a bunch of variables which we will realistically set only once
and never again. Because of that, and to save up on namespace, we
will write these in uppercase and type them as `Final`.
## Introspection & debugging
First are the two debugging variables, `DEBUG` and `EXAMPLE`.
`DEBUG` should control behavior checks.
`EXAMPLE` should control examples which are not necessary for the project.
<code>
DEBUG: Final[bool] = False
EXAMPLE: Final[bool] = True
</code>
and we need a simple way to output something only when `EXAMPLE` is
`True`, so we define the function `example`
<code>
def example(x):
if EXAMPLE:
return x
</code>
## Paths
Then we have our data directory, `DATA_DIR`, which contains the data
we cache from `allensdk`.
<code>
DATA_DIR: Final[pathlib.Path] = pathlib.Path('./data/')
</code>
and we need to ensure that this directory exists
<code>
DATA_DIR.mkdir(parents=True, exist_ok=True)
</code>
Furthermore, we have the path to the manifest file, again relative to
the project root, which is by default the `manifest.json` file located
directly in `DATA_DIR`.
<code>
MANIFEST_PATH: Final[pathlib.Path] = DATA_DIR / 'manifest.json'
</code>
## Cache & sessions
Next is our `allensdk` cache and the sessions in it.
First define the type aliases `Cache` and `Session` for easier
typing in functions.
<code>
Cache: TypeAlias = EcephysProjectCache
Session: TypeAlias = EcephysSession
</code>
Set the cache's timeout, `CACHE_TIMEOUT`, which is measured in
seconds:
<code>
CACHE_TIMEOUT: Final[int] = 30*60 # 30 minutes
</code>
Define the global cache, `CACHE`,
<code>
CACHE: Final[Cache] = EcephysProjectCache.from_warehouse(manifest=MANIFEST_PATH, timeout=CACHE_TIMEOUT)
</code>
Load the table of sessions for simple access.
<code>
SESSIONS_TABLE: Final = CACHE.get_session_table()
example(SESSIONS_TABLE.head())
</code>
Get the complete list of session ids.
<code>
SESSION_IDS: Final[Sequence[int]] = SESSIONS_TABLE.index
example(SESSION_IDS)
</code>
We typically want to work on just one session, `CURRENT_SESSION`.
That can be specified here for convenience using `CURRENT_SESSION_ID`.
Functions strive to accept a `session` parameter where meaningful and
default to `CURRENT_SESSION`.
<code>
%%capture
CURRENT_SESSION_ID: Final[int] = 715093703
assert CURRENT_SESSION_ID in SESSION_IDS
CURRENT_SESSION: Final[Session] = CACHE.get_session_data(CURRENT_SESSION_ID)
example(CURRENT_SESSION.metadata)
</code>
# Accessing filtered data
The `Session` object exposes a lot of useful functions and properties
to us. In order to free us from having to always type the session
(which will overwhelmingly be `CURRENT_SESSION`) and to simultaneously
allow us to filter our data without having to go through much trouble,
we will define simple wrappers around those functions.
Typically, these functions accept a `session` parameter which defaults
to `CURRENT_SESSION`, and a bunch of keyword arguments which filter
the resulting dataset.
Each argument `foo=bar`, unless stated otherwise, filters the `foo`
column of the resulting dataset to values matching `bar`. See the
commentary at the start of `constraints.py` for a full description of
what values `bar` can take.
<code>
def get_sessions(**kwargs):
"""Return a table of the matching sessions.
The following filters are meaningful:
- published_at (time)
- specimen_id (integer, key)
- session_type ('brain_observatory_1.1' or 'functional_connectivity')
- age_in_days (float)
- sex ('M' or 'F')
- full_genotype (string)
- unit_count (integer)
- channel_count (integer)
- probe_count (integer)
- ecephys_structure_acronyms (list of strings)
If an argument `__total__=False` is passed, additional filters may
be provided with no effect on the result.
"""
return filter_df(SESSIONS_TABLE, FIELD(**kwargs))
def get_session_ids(**kwargs):
"""Return the matching session ids.
See `get_sessions` for the list of meaningful filters.
"""
return get_sessions(**kwargs).index
example(get_sessions(sex='M', unit_count=RANGE(650, None)))
</code>
<code>
def get_units(ecephys_structure_acronym = None,
unit_ids = None,
session: Session = CURRENT_SESSION,
**kwargs):
"""Return a `Session.units` dataframe of the matching units in `session`.
The `unit_ids` argument narrows the stimulus presentations
considered to those whose id it contains.
The following filters are meaningful:
- waveform_PT_ratio (float)
- waveform_amplitude (float)
- amplitude_cutoff (float)
- cluster_id (integer, key)
- cumulative_drift (float)
- d_prime (float or null)
- firing_rate (float)
- isi_violations (float)
- isolation_distance (float or null)
- L_ratio (float or null)
- local_index (integer)
- max_drift (float)
- nn_hit_rate (float or null)
- nn_miss_rate (float or null)
- peak_channel_id (integer, key)
- presence_ratio (float)
- waveform_recovery_slope (float or null)
- waveform_repolarization_slope (float)
- silhouette_score (float or null)
- snr (float)
- waveform_spread (float)
- waveform_velocity_above (float or null)
- waveform_velocity_below (float or null)
- waveform_duration (float)
- filtering (string)
- probe_channel_number (integer)
- probe_horizontal_position (integer)
- probe_id (integer)
- probe_vertical_position (integer)
- structure_acronym (string)
- ecephys_structure_id (float, key)
- ecephys_structure_acronym (string)
- anterior_posterior_ccf_coordinate (float or null)
- dorsal_ventral_ccf_coordinate (float or null)
- left_right_ccf_coordinate (float or null)
- probe_description (string, probeA..F)
- location (object)
- probe_sampling_rate (float)
- probe_lfp_sampling_rate (float)
- probe_has_lfp_data (bool)
If an argument `__total__=False` is passed, additional filters may
be provided with no effect on the result.
"""
if ecephys_structure_acronym is not None:
kwargs['ecephys_structure_acronym'] = ecephys_structure_acronym
units = session.units
if unit_ids is not None:
units = units.loc[unit_ids]
return filter_df(units, FIELD(**kwargs))
def get_unit_ids(*args, **kwargs):
"""Return the matching unit ids in `session`.
See `get_units` for the list of meaningful filters.
"""
return get_units(*args, **kwargs).index
example(get_units(isi_violations = RANGE(None, 0.7),
structure_acronym = 'VISam').head())
</code>
<code>
def get_stimulus_presentations(stimulus_name = None,
stimulus_presentation_ids = None,
stimulus_condition_id = None,
session: Session = CURRENT_SESSION,
**kwargs):
"""Return the Sessions.stimulus_presentations dataframe of `session`.
The `stimulus_presentation_ids` argument narrows the stimulus
presentations considered to those whose id it contains.
The following filters are meaningful:
- stimulus_block (float or null, key)
- start_time (float)
- stop_time (float)
- contrast (float or null)
- spatial_frequency (float, string, or null)
- frame (float or null)
- stimulus_name (string)
- x_position (float or null)
- y_position (float or null)
- orientation (float or null)
- temporal_frequency (float or null)
- size (object)
- color (-1.0, 1.0, or null)
- phase (object)
- duration (float)
- stimulus_condition_id (integer, key)
If an argument `__total__=False` is passed, additional filters may
be provided with no effect on the result.
"""
if stimulus_name is not None:
kwargs['stimulus_name'] = stimulus_name
if stimulus_condition_id is not None:
kwargs['stimulus_condition_id'] = stimulus_condition_id
stimulus_presentations = session.stimulus_presentations
if stimulus_presentation_ids is not None:
stimulus_presentations = stimulus_presentations.loc[stimulus_presentation_ids]
return filter_df(stimulus_presentations, FIELD(**kwargs))
def get_stimulus_presentation_ids(*args, **kwargs):
"""Return the matching stimulus presentation ids in `session`.
See `get_stimulus_presentations` for a list of meaningful filters.
"""
return get_stimulus_presentations(*args, **kwargs).index
example(get_stimulus_presentations(stimulus_name = 'static_gratings',
orientation = AND(NOT('null'),
RANGE(30, 60, ub_strict=False))))
</code>
<code>
def get_presentationwise_spike_times(session: Session = CURRENT_SESSION, **kwargs):
"""Return a table of the spike times of the matching units and stimuli.
All filters which `get_units` and `get_stimulus_presentations`
accept are meaningful.
"""
kwargs['__total__'] = False
return session.presentationwise_spike_times(
stimulus_presentation_ids = get_stimulus_presentation_ids(session = session, **kwargs),
unit_ids = get_unit_ids(session = session, **kwargs)
)
example(get_presentationwise_spike_times(stimulus_name = 'static_gratings',
orientation = 0.0,
ecephys_structure_acronym = 'VISam'))
</code>
<code>
def get_conditionwise_spike_statistics(use_rates: Optional[bool] = False,
session: Session = CURRENT_SESSION,
**kwargs):
"""Return a table of the spike statistics for each stiulus condition.
If `use_rates` is True, use firing rates, otherwise use spike counts.
All filters which `get_units` and `get_stimulus_presentations`
accept are meaningful.
"""
kwargs['__total__'] = False
return session.conditionwise_spike_statistics(
stimulus_presentation_ids = get_stimulus_presentation_ids(session = session, **kwargs),
unit_ids = get_unit_ids(session = session, **kwargs),
use_rates = use_rates
)
example(get_conditionwise_spike_statistics(ecephys_structure_acronym = 'VISam',
stimulus_name = OR('static_gratings','drifting_gratings')))
</code>
<code>
def get_annotated_spike_times(session: Session = CURRENT_SESSION, **kwargs):
"""Return a table of spike times alongside the unit and stimulus information.
All filters which `get_units` and `get_stimulus_presentations`
accept are meaningful.
"""
kwargs['session'] = session
kwargs['__total__'] = False
return get_presentationwise_spike_times(**kwargs) \
.merge(get_stimulus_presentations(**kwargs), left_on='stimulus_presentation_id', right_index=True) \
.merge(get_units(**kwargs), left_on='unit_id', right_index=True)
example(get_annotated_spike_times(stimulus_name=ISIN(['static_gratings', 'drifting_gratings']),
ecephys_structure_acronym=ISIN(['VISp', 'DG', 'LGd', 'APN'])))
</code>
|
{
"filename": "00_Setup_1.ipynb",
"repository": "tebe-nigrelli/MMN-Group-Project",
"query": "transformed_from_existing",
"size": 127400,
"sha": ""
}
|
# qc_rna-10xv3.ipynb
Repository: pachterlab/voyagerpy
# Basic quality control on scRNA-seq data wih 10X v3
<code>
!git clone https://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@github.com/cellatlas/cellatlas.git > /dev/null
!pip install --quiet git+https://github.com/pmelsted/voyagerpy
</code>
<code>
!pip install --quiet anndata
</code>
<code>
!gunzip /content/cellatlas/examples/rna-10xv3/cellatlas_out/adata.h5ad.gz
</code>
<code>
import voyagerpy as vp
import anndata
from matplotlib import pyplot as plt
import numpy as np
plt.rcParams.update({
'figure.dpi': 120,
'font.size': 20})
_ = plt.ion()
%config InlineBackend.figure_format = 'retina'
</code>
<code>
adata = anndata.read_h5ad("/content/cellatlas/examples/rna-10xv3/cellatlas_out/adata.h5ad")
</code>
<code>
adata.var
</code>
<code>
is_mt = adata.var['gene_name'].str.contains('^mt-', case=False).values
vp.utils.add_per_cell_qcmetrics(adata, subsets={'mito': is_mt})
adata.obs.head()
</code>
<code>
adata = adata[adata.obs["subsets_mito_percent"].notna()].copy()
</code>
<code>
(adata.obs["subsets_mito_percent"]>0).sum()
</code>
<code>
qc_features = ["sum", "detected", "subsets_mito_percent"]
_ = vp.plt.plot_barcode_data(
adata,
y=qc_features,
ncol=3,
)
</code>
<code>
_ = vp.plt.plot_barcodes_bin2d(adata, x='sum', y='detected', cmin=1)
</code>
<code>
_ = vp.plt.plot_barcodes_bin2d(adata, x='sum', y='subsets_mito_percent', cmin=1)
</code>
<code>
cells_to_keep = adata.obs["subsets_mito_percent"] < 20
_, genes_to_keep = np.where(adata[cells_to_keep, :].X.sum(axis=0) > 0)
adata = adata[cells_to_keep, genes_to_keep].copy()
adata
</code>
|
{
"filename": "qc_rna-10xv3.ipynb",
"repository": "pachterlab/voyagerpy",
"query": "transformed_from_existing",
"size": 330759,
"sha": ""
}
|
# llmops_with_langsmith_1.ipynb
Repository: buzzbing/llmops-platforms
### Configuration Requirements:
Definition of Environment Variables
- OPENAI_API_KEY= [openai api key]
- OPENAI_ORGANIZATION=[organization key]
- LANGCHAIN_TRACING_V2=true
- LANGCHAIN_ENDPOINT="https://api.smith.langchain.com"
- LANGCHAIN_API_KEY=[ langsmith apikey]
- LANGCHAIN_PROJECT=[project name]
<code>
# !pip install -U langchain langsmith
</code>
<code>
from dotenv import find_dotenv,load_dotenv
_ = load_dotenv(find_dotenv())
</code>
### Tracing
- A trace typically represents a single request or operation. It contains overall input and output of the function and metadata about the request, such as the user, the session, duration of units of work, token usage, costs and tags. (link)
- It is essentially a series of steps that an application takes to go from input to output.
### Run
- Each of these individual steps in a trace is represented by a Run.
- This could be anything from single call to an LLM or chain, to a prompt formatting call, to a runnable lambda invocation.
### Project
- A collection of traces
- It can be taken as a container for all the traces that are related to a single application or service
#### Trace Example 1
<code>
import openai
from langsmith.wrappers import wrap_openai
from langsmith import traceable
client = wrap_openai(openai.Client())
@traceable
def get_response(user_input: str):
result = client.chat.completions.create(
messages=[{"role": "user", "content": user_input}],
model="gpt-3.5-turbo"
)
return result.choices[0].message.content
response = get_response("What is LLMops in Machine Learning? What are the best tools for LLMops operation?")
</code>
<code>
print(response)
</code>
#### Trace Example 2
<code>
import openai
from langsmith import traceable
client = wrap_openai(openai.Client())
@traceable
def openai_response(system_prompt: str, user_input: str):
result = client.chat.completions.create(
messages=[{"role": "system", "content": system_prompt},
{"role": "user","content":user_input}],
model="gpt-3.5-turbo",
temperature=0.5,
frequency_penalty=0.1,
timeout=5,
)
return result.choices[0].message.content
</code>
<code>
system_prompt = '''You are an eloquent travel assistant. Your task is to recommend best cities for a vacation to users based on the requirements of the user. Understand the requirements and also recommend the things users need to carry with them to travel to the given cities.'''
user_prompt = '''I want to travel in February. The city must not be freezing cold neither too hot to travel. Asian Cities will be preferred.'''
response = openai_response(system_prompt,user_prompt)
</code>
<code>
print(response)
</code>
#### Trace Example 3
<code>
from typing import List
from langchain_openai import ChatOpenAI
from langchain.prompts import PromptTemplate
from langchain_core.output_parsers import JsonOutputParser
model = ChatOpenAI(temperature=0)
joke_query = "Tell me a joke."
parser = JsonOutputParser()
prompt = PromptTemplate(
template="Answer the user query.\n{format_instructions}\n{query}\n",
input_variables=["query"],
partial_variables={"format_instructions": parser.get_format_instructions()},
)
chain = prompt | model | parser
chain.invoke({"query": joke_query})
</code>
### Dataset Creation
- Dataset can be created by collecting the prompt input and output from debugging or other runs.
- The dataset can be modified by updating the output values if desired.
- We can also create dataset by uploading a csv file with inputs and outputs as key-value pairs.
- Dataset of traces including all the metadata of each run can also be created
- Datasets are created to evaluate the responses from same model or different models on different evaluation criteria.
Methods of Dataset Creation:
1. Importing CSV files
2. Through Existing Runs
<code>
from langsmith import Client
example_inputs = [
("What is the largest mammal?", "The blue whale"),
("What do mammals and birds have in common?", "They are both warm-blooded"),
("What are reptiles known for?", "Having scales"),
("What's the main characteristic of amphibians?", "They live both in water and on land"),
]
client = Client()
dataset_name = "Elementary Animal Questions"
dataset = client.create_dataset(
dataset_name=dataset_name, description="Questions and answers about animal phylogenetics.",
)
for input_prompt, output_answer in example_inputs:
client.create_example(
inputs={"question": input_prompt},
outputs={"answer": output_answer},
metadata={"source": "Wikipedia"},
dataset_id=dataset.id,
)
</code>
<code>
import os
from langsmith import Client
client = Client()
dataset_name = "run1"
runs = client.list_runs(
project_name=os.environ.get('LANGCHAIN_PROJECT'),
execution_order=1,
error=False,
)
dataset = client.create_dataset(dataset_name, description="Dataset creation using Runs")
for run in runs:
client.create_example(
inputs=run.inputs,
outputs=run.outputs,
dataset_id=dataset.id,
)
</code>
### Evaluation
- Evaluator is a function that takes in a set of inputs and outputs from your chain, agent, or model, and returns a score (or multiple scores)
- The common automated evaluator types are:
- Simple Heuristics: Checking for regex matches, presence/absence of certain words or code, etc.
- AI-assisted: Instruct an "LLM-as-judge" to grade the output of a run based on the prediction and reference answer (or retrieved context).
- When running an evaluation, example inputs are run through Task to produce Runs, which are then passed into evaluator along with the Example. The function then returns an Evaluation Result, which specifies your metric name and score.
#### Evaluation by Comparison
<code>
from langsmith import Client
from langchain_openai import chat_models
from langchain import prompts, smith
from langchain.schema import output_parser
# Define your runnable or chain below.
system_prompt = """You are an eloquent travel assistant. Your task is to recommend best cities for a vacation to users based on the requirements of the user. Understand the requirements and also recommend the things users need to carry with them to travel to the given cities."""
user_prompt = """I want to travel in February. The city must not be freezing cold neither too hot to travel. Asian Cities will be preferred."""
prompt = prompts.ChatPromptTemplate.from_messages(
[("system", system_prompt), ("human", "{user_input}")]
)
llm = chat_models.ChatOpenAI(model="gpt-4", temperature=0)
chain = prompt | llm | output_parser.StrOutputParser()
client = Client()
results = client.run_on_dataset(
dataset_name="langsmith_dataset",
llm_or_chain_factory=chain,
project_name="langsmith_eval5",
)
</code>
#### Evaluation by LangSmith Evaluators
##### Evaluation in Chat model
<code>
examples = [
{
"inputs": {
"question": "How does that apply?",
"chat_history": [
{
"role": "user",
"content": "I’m trying tto uunderstand instein’s theory.",
},
{
"role": "assistant",
"content": "Which one? He's known for several theories.",
},
{"role": "user", "content": "Thee one about time and spae."},
{
"role": "assistant",
"content": "Ah, you're referring to the theory of relativity. There are two parts: special and general. Which one?",
},
],
},
"outputs": {
"expected": "Special relativity, proposed by Einstein in 1905, deals with objects in uniform motion, especially those moving at the speed of light. It introduced the idea that time and space are relative and can change in relation to each other. For instance, time can appear to move slower for an object moving close to the speed of light."
},
},
{
"inputs": {
"question": "What's the main difference?",
"chat_history": [
{"role": "user", "content": "Can yyou contrast DNA and RNA for me?"},
{
"role": "assistant",
"content": "Certainly. DNA and RNA are both nucleic acids but have different roles, structures, and properties. Do you want specifics?",
},
],
},
"outputs": {
"expected": "The main structural differences between DNA and RNA include: 1) DNA is double-stranded while RNA is single-stranded. 2) The sugar in the backbone of RNA is ribose, whereas in DNA it's deoxyribose. 3) DNA uses the bases adenine (A), cytosine (C), guanine (G), and thymine (T); RNA uses adenine (A), cytosine (C), guanine (G), and uracil (U) instead of thymine."
},
},
{
"inputs": {
"question": "what led them to such a draastic action?",
"chat_history": [
{"role": "user", "content": "tell me about the Boston Tea Party."},
{
"role": "assistant",
"content": "The Boston Tea Party was a political protest by the American colonists against the British government in 1773. They were protesting the Tea Act, which allowed the British East India Company to sell tea directly to the colonies, bypassing colonial merchants.",
},
],
},
"outputs": {
"expected": "The colonists undertook the Boston Tea Party as a drastic action due to multiple reasons: 1) They believed the Tea Act was a violation of their rights as Englishmen, as they were being taxed without their consent. 2) The act gave the British East India Company a monopoly on tea sales, threatening local businesses. 3) The act was seen as another example of the British government's increasing interference in colonial affairs. The protest was a way to show their strong opposition to British policies."
},
},
{
"inputs": {
"question": "thats a scary one. can it be avoideed?",
"chat_history": [
{"role": "user", "content": "I'm learning bout genetic disorders."},
{
"role": "assistant",
"content": "Genetic disorders are diseases caused by abnormalities in an individual's DNA. They can be inherited or result from mutations. One common one is Huntington's disease.",
},
],
},
"outputs": {
"expected": "Huntington's disease is a hereditary genetic disorder caused by a mutation in the HTT gene. If a person inherits the defective gene, they will eventually develop the disease. Currently, there's no cure for Huntington's, but its onset can be delayed with treatment. Genetic counseling and testing can help prospective parents understand the risks of passing the mutation to their offspring."
},
},
{
"inputs": {
"question": "Which one?",
"chat_history": [
{
"role": "user",
"content": "I'm confused aboutt stars. what even aaaare they?",
},
{
"role": "assistant",
"content": "Stars are celestial bodies made mostly of hydrogen and helium. They generate light and heat through nuclear fusion in their cores.",
},
{
"role": "user",
"content": "there''s a classification based on theirbrightness, right?",
},
{
"role": "assistant",
"content": "Yes",
},
],
},
"outputs": {
"expected": "Yes, stars are classified based on their brightness using a system called the Hertzsprung-Russell (H-R) diagram. In this diagram, stars are categorized into main-sequence stars, giants, supergiants, and white dwarfs, based on their luminosity and temperature. The Sun, for instance, is a main-sequence star."
},
},
]
</code>
<code>
from langsmith import Client
import uuid
client = Client()
dataset_name = f"Chat Bot Evals Single-Turn Example - {uuid.uuid4()}"
dataset = client.create_dataset(dataset_name)
client.create_examples(
inputs=[e["inputs"] for e in examples],
outputs=[e["outputs"] for e in examples],
dataset_id=dataset.id,
)
</code>
<code>
from langchain.prompts import ChatPromptTemplate, MessagesPlaceholder
from langchain.schema.output_parser import StrOutputParser
from langchain_openai import ChatOpenAI
# An example chain
chain = (
ChatPromptTemplate.from_messages(
[
("system", "You are a helpful tutor AI."),
MessagesPlaceholder(variable_name="chat_history"),
("user", "{input}"),
]
)
| ChatOpenAI(model="gpt-3.5-turbo")
| StrOutputParser()
)
</code>
<code>
from langchain.adapters.openai import convert_openai_messages
from langsmith.evaluation import LangChainStringEvaluator, evaluate
from langsmith.schemas import Example, Run
def predict(inputs: dict):
return chain.invoke(
{
"input": inputs["question"],
"chat_history": convert_openai_messages(inputs["chat_history"]),
}
)
def format_evaluator_inputs(run: Run, example: Example):
return {
"input": example.inputs["question"],
"prediction": next(iter(run.outputs.values())),
"reference": example.outputs["expected"],
}
correctness_evaluator = LangChainStringEvaluator(
"labeled_score_string",
config={"criteria": "correctness", "normalize_by": 10},
prepare_data=format_evaluator_inputs,
)
results = evaluate(
predict,
data=dataset_name,
experiment_prefix="Chat Single Turn",
evaluators=[correctness_evaluator],
metadata={"model": "gpt-3.5-turbo"},
)
</code>
##### Evaluation from Run Dataset
<code>
from langsmith import Client
from langchain_openai.chat_models import ChatOpenAI
from langchain.smith import RunEvalConfig, run_on_dataset
from langchain import prompts,smith
from langchain.schema import output_parser
system_prompt = '''You are an eloquent travel assistant. Your task is to recommend best cities for a vacation to users based on the requirements of the user. Understand the requirements and also recommend the things users need to carry with them to travel to the given cities.'''
user_prompt = '''I want to travel in February. The city must not be freezing cold neither too hot to travel. Asian Cities will be preferred.'''
prompt = prompts.ChatPromptTemplate.from_messages(
[
("system", system_prompt),
("human", "{user_input}")
]
)
llm = ChatOpenAI(model="gpt-3.5-turbo", temperature=0)
chain = prompt | llm | output_parser.StrOutputParser()
evaluation_config = RunEvalConfig(
evaluators=[
"qa",
"context_qa",
"cot_qa",
],
reference_key="output",
input_key="user_input",
)
client = Client()
run_on_dataset(
client=client,
dataset_name="langsmith_dataset",
llm_or_chain_factory=chain,
evaluation=evaluation_config,
)
</code>
### Threads
Traces which are part of the same conversation can be tracked using Threads. A metadata key is attached to each trace
<code>
q1 = "What is LLMOps in Machine Learning? What are the best tools for LLMOps operation?"
q2 = "What is the difference between MLOps and LLMOps in Machine Learning"
</code>
<code>
import openai
from langsmith import traceable
import uuid
client = openai.Client()
# define session id
session_id = str(uuid.uuid4())
# define function for call
@traceable(
run_type="chain",
name="OpenAI Assistant",
)
def assistant(messages: list[dict]):
return (
client.chat.completions.create(
model="gpt-3.5-turbo",
messages=[{"role": "system", "content": "You are a Machine Learning assistant specialized in MLOps."}]
+ messages,
)
.choices[0]
.message
)
messages = [{"role": "user", "content": q1}]
response = assistant(messages, langsmith_extra={"metadata": {"session_id": session_id}})
messages = messages + [response, {"role": "user", "content": q2}]
response = assistant(messages, langsmith_extra={"metadata": {"session_id": session_id}})
</code>
|
{
"filename": "llmops_with_langsmith_1.ipynb",
"repository": "buzzbing/llmops-platforms",
"query": "transformed_from_existing",
"size": 26285,
"sha": ""
}
|
# rds2h5ad_1.ipynb
Repository: jiang-junyao/DRCTdb
<code>
import scanpy as sc
</code>
<code>
sample1 = sc.read_h5ad('../../data/scATAC-seq/Sample1/Rds/sample1_scATAC-seq_80k_processed.h5ad')
</code>
<code>
print(sample1)
len(sample1.obs['cell_type'].value_counts())
</code>
<code>
sample4 = sc.read_h5ad('../../data/scATAC-seq/Sample4/sample4_scATAC-seq_30k_processed.h5ad')
</code>
<code>
print(sample4)
len(sample4.obs['cell_type'].value_counts())
</code>
<code>
sample7 = sc.read_h5ad('../../data/scATAC-seq/Sample7/sample7_scATAC-seq_50k_processed.h5ad')
</code>
<code>
print(sample7)
len(sample7.obs['cell_type'].value_counts())
</code>
<code>
sample8 = sc.read_h5ad('../../data/scATAC-seq/Sample8/sample8_scATAC-seq_100k_processed.h5ad')
</code>
<code>
print(sample8)
len(sample8.obs['cell_type'].value_counts())
</code>
<code>
sample10 = sc.read_h5ad('../../data/scATAC-seq/Sample10/sample10_scATAC-seq_950k_processed.h5ad')
</code>
<code>
print(sample10)
len(sample10.obs['cell_type'].value_counts())
</code>
<code>
sample11 = sc.read_h5ad('../../data/scATAC-seq/Sample11/sample11_scATAC-seq_17k_processed.h5ad')
</code>
<code>
print(sample11)
len(sample11.obs['cell_type'].value_counts())
</code>
<code>
sample12 = sc.read_h5ad('../../data/scATAC-seq/Sample12/sample12_scATAC-seq_104k_processed.h5ad')
</code>
<code>
print(sample12)
len(sample12.obs['cell_type'].value_counts())
</code>
<code>
sample13 = sc.read_h5ad('../../data/scATAC-seq/Sample13/sample13_scATAC-seq_4k_processed.h5ad')
</code>
<code>
print(sample13)
len(sample13.obs['cell_type'].value_counts())
</code>
<code>
sample14 = sc.read_h5ad('../../data/scATAC-seq/Sample14/sample14_scATAC-seq_95k_processed.h5ad')
</code>
<code>
print(sample14)
len(sample14.obs['cell_type'].value_counts())
</code>
<code>
sample16 = sc.read_h5ad('../../data/scATAC-seq/Sample16/sample16_scATAC-seq_35k_processed.h5ad')
</code>
<code>
print(sample16)
len(sample16.obs['cell_type'].value_counts())
</code>
<code>
sample23 = sc.read_h5ad('../../data/scATAC-seq/Sample23/sample23_scATAC-seq_95k_processed.h5ad')
</code>
<code>
print(sample23)
len(sample23.obs['cell_type'].value_counts())
</code>
<code>
sample24 = sc.read_h5ad('../../data/scATAC-seq/Sample24/sample24_scATAC-seq_52k_processed.h5ad')
</code>
<code>
print(sample24)
len(sample24.obs['cell_type'].value_counts())
</code>
<code>
sample26 = sc.read_h5ad('../../data/scATAC-seq/Sample26/Sample26_scATAC-seq_229k_processed.h5ad')
</code>
<code>
print(sample26)
len(sample26.obs['cell_type'].value_counts())
</code>
<code>
sample27 = sc.read_h5ad('../../data/scATAC-seq/Sample27/Sample27_scATAC-seq_133k_processed.h5ad')
</code>
<code>
print(sample27)
len(sample27.obs['cell_type'].value_counts())
</code>
<code>
sample3 = sc.read_h5ad('../../data/scATAC-seq/Sample3/sample3_scATAC-seq_756k_processed.h5ad')
</code>
<code>
print(sample3)
len(sample3.obs['cell_type'].value_counts())
</code>
<code>
sample5 = sc.read_h5ad('../../data/h5ad/sample5_scATAC-seq_720k_processed.h5ad')
</code>
<code>
print(sample5)
len(sample5.obs['cell_type'].value_counts())
</code>
|
{
"filename": "rds2h5ad_1.ipynb",
"repository": "jiang-junyao/DRCTdb",
"query": "transformed_from_existing",
"size": 21542,
"sha": ""
}
|
# graph-prac.ipynb
Repository: kortschak/graphprac
# Graphical analysis of protein interactions in yeast
The aim of this practical is to examine some uses of graphical analysis in a biological setting. The analyses are identical to those demonstrated in the London Tube Graph examples in the introduction to systems biology lecture.
The approaches we saw in the London Tube Map example involved visual examination of the coloured nodes and edges of the graph. For large biological (or other) graphs, this is not feasible. The graph we will be looking at today is shown here.

Questions to be answered for the assignment are written in **bold**. Answers are to be written in the blank cells immediately below the question and the assignment should be submitted as a Notebook file with the name `graph-prac-aNNNNNNN.ipynb` where aNNNNNNN is your a-number (jupyter: File → Download as → Notebook (.ipynb) when you have written your answers). This file should be packaged into a zip file called `graph-prac-aNNNNNNN.zip` with no other files. A cover sheet will not be necessary for submission. Submit via MyUni.
The assignment is due Monday 27nd October before 5pm ACDT.
### Programming language
The practical uses the [Go programming language](https://golang.org/). Go is a statically typed, compiled language, but we will be using it through an interactive environment called Jupyter. This tries to make Go behave as an interpretted language, and this may cause some problems at some stages. If you have any problems, please ask the demonstrators for help.
### Setting up the package imports
Go modularises code into packages (much the same way as other languages, though different languages will use different terms for the same concept). Go packages must be imported before they can be used. In a compiled Go program this *must* happen at the beginning of a source code file, though in jupyter, this is not strictly necessary.
The packages that we will use are for printing, `"fmt"`, logging errors, `"log"`, and performing the graph analyses, `"github.com/kortschak/graphprac"`. The first two packages are provided with the language and the last is a small package that wraps graph routines made available through the graph packages of the https://gonum.org project, [`graph/...`](https://godoc.org/gonum.org/v1/gonum/graph/).
<code>
import (
"fmt"
"log"
"github.com/kortschak/graphprac"
)
</code>
The documentation for the `"github.com/kortschak/graphprac"` package is available from the godoc.org website: http://godoc.org/github.com/kortschak/graphprac. You can click on links on the documentation page to see the source of the functions and to link to the definitions of types and externally provided functions. This is an excellent way to get to understand the code.
The routines wrapped by the `"github.com/kortschak/graphprac"` are in [`graph/network`](https://godoc.org/gonum.org/v1/gonum/graph/network) and [`graph/community`](https://godoc.org/gonum.org/v1/gonum/graph/community).
### Read in a graph
The graph is from a [data set](http://vlado.fmf.uni-lj.si/pub/networks/data/bio/Yeast/Yeast.htm) used in an analysis of topological structure in the yeast protein interaction network (DOI:[10.1093/nar/gkg340](https://doi.org/10.1093/nar/gkg340)).
<code>
infile := "YeastL.dot"
</code>
<code>
g, err := graphprac.NewGraph(infile)
if err != nil {
log.Fatalf("failed to read graph: %v", err)
}
{
fmt.Printf("G has %d nodes and %d edges.\n", g.Nodes().Len(), g.Edges().Len())
}
</code>
**Notice that the number of nodes agrees with the data set summary linked above, but the number of edges disagrees. Suggest why this might be.**
### Examining the nodes
There is no `head` function in Go (we could write one, but the code is short, so there is no need).
The following code loops over the first 10 elements of the nodes slice (essentially an array - there are differences in Go, but that is not important here) and prints out the node value stored in `n`.
<code>
for _, n := range graphprac.NodesOf(g)[:10] {
fmt.Println(n)
}
</code>
Note that running this multiple times will result in different sets of nodes being printed as the nodes returned by `g.Nodes()` are selected in a random order, so the first ten will differ. Try this out.
**Why is it not important that a node list be returned in a specific order?**
### Network analysis
We are going to look at nodes that have a high connectivity or potential for information flow through the network.
Two measures that we can use to do this (*very roughly*) are node betweenness centrality and PageRank.
The routines provided in the `graphprac` package write their analysis results into the graph that is provided as a parameter. This is not how we normally do this kind of analysis, but it makes the practical simpler.
The writing into the graph is done via a set of attributes that have names that can be queried using the functions in the package.
**How can an attribute be queried?**
**Use an example to demonstrate.**
The first analysis is for node betweenness of G.
<code>
graphprac.Betweenness(g)
</code>
<code>
nodes, err := graphprac.NodesByAttribute("betweenness", g)
if err != nil {
log.Fatalf("failed to obtain nodes: %v", err)
}
for _, n := range nodes[:10] {
attr := n.Attributes
fmt.Printf("%s %s -- %s\n", n.Name, attr.Get("betweenness"), attr.Get("desc"))
}
</code>
**If you rerun the code block above, does the order/set of nodes change like in the previous example? Why?**
Next we perform a PageRank analysis of G. There are two additional parameters here, `damp` and `tol`.
**What is the purpose of these two parameters? _Hint: You will need to read about PageRank._**
<code>
graphprac.PageRank(g, 0.85, 1e-4)
</code>
Now we print out the ten highest ranked nodes and keep the highest ranked node of all.
<code>
nodes, err := graphprac.NodesByAttribute("rank", g)
if err != nil {
log.Fatalf("failed to obtain nodes: %v", err)
}
bestRank := nodes[0]
for _, n := range nodes[:10] {
attr := n.Attributes
fmt.Printf("%s %s -- %s\n", n.Name, attr.Get("rank"), attr.Get("desc"))
}
</code>
**Look at the two sets of highest ranked nodes from the betweenness and PageRank analyses. How well do they agree? How does this situation compare to the case of the London Tube Graph example? Why do you think this is?**
The next step is to identify sets of nodes that interact more strongly within the set than they do between sets. These sets are called communities.

The `graphprac.Communities` function takes a single extra parameter, `resolution`. We are using a resolution of 5.
<code>
graphprac.Communities(g, 5)
</code>
Now we are going to identify the community that the highest PageRanked node is in. Note that the algorithm used to identify communities is a randomised algorithm, the [Louvain Algorithm](https://en.wikipedia.org/wiki/Louvain_Modularity), and so different runs will produce a different name for the community and may include slightly different community memberships.
*Community detection is an NP-hard problem and the Louvain Algorithm gives us a reasonable approximation in reasonable time.*
<code>
nodes, err := graphprac.NodesByAttribute("community", g)
if err != nil {
log.Fatalf("failed to get community: %v", err)
}
comm := ""
for _, n := range nodes {
attr := n.Attributes
if n.Name == bestRank.Name {
comm = attr.Get("community")
break
}
}
{
fmt.Printf("%s is in community %s\n", bestRank.Name, comm)
}
</code>
<code>
for _, n := range nodes {
attr := n.Attributes
if attr.Get("community") == comm {
fmt.Printf("%s -- %s\n", n.Name, attr.Get("desc"))
}
}
</code>
**What are the functions of these proteins? Would you expect them to be in the same community? Hint:http://www.yeastgenome.org/**
**What happens when you alter the resolution parameter to `graphprac.Communities`? Note here that there is another tool, Gephi, that performs community detection but inverts the meaning of the resolution parameter. Do not use their definition.**
We can render the graph visually using external tools provided by [GraphViz](https://www.graphviz.org/). The `graphprac.Induce` function produces a subgraph based on `g` that only includes nodes in `g` that are also in the `by` parameter (this is termed an [induced subgraph](https://en.wikipedia.org/wiki/Induced_subgraph)). `graphprac.Draw` then generates an SVG graphic rendering of the graph.
<code>
by := []*graphprac.Node{}
for _, n := range nodes {
attr := n.Attributes
if attr.Get("community") == comm {
by = append(by, n)
}
}
subgraph := graphprac.Induce(g, by)
svg, err := graphprac.Draw(subgraph, "fdp")
if err != nil {
fmt.Println(err)
}
display.SVG(svg)
</code>
Finally we are going to look for potential targets to disrupt the function of this community. By looking for edges that have a high edge betweenness we may be able to identify candidates for molecular disruption.
**Run the two code cells below and choose an interaction pair that looks like it might be a good candidate for a druggable target. Write the names of the two proteins you chose in the next cell.**
**Notice that the number of edges reported below is significantly higher than shown in the graph rendering above. Have a look at the source code associated with [`graphprac.Induce`](https://godoc.org/github.com/kortschak/graphprac#Induce) and explain why this is the case. Why might it be useful to be more inclusive in the case here of looking for druggable targets?**
<code>
graphprac.EdgeBetweenness(g)
</code>
<code>
edges, err := graphprac.EdgesByAttribute("edge_betweenness", g)
if err != nil {
log.Fatalf("failed to obtain nodes: %v", err)
}
for _, e := range edges {
fattr := e.F.Attributes
tattr := e.T.Attributes
if fattr.Get("community") != comm && tattr.Get("community") != comm {
continue
}
attr := e.Attributes
fmt.Printf("%s--%s %s (%s--%s)\n", e.F.Name, e.T.Name, attr.Get("edge_betweenness"), fattr.Get("desc"), tattr.Get("desc"))
}
</code>
**Write a page (~300 words) explaining why you chose the target you did and how it could potentially be investigated further. Give references for information you introduce.**
|
{
"filename": "graph-prac.ipynb",
"repository": "kortschak/graphprac",
"query": "transformed_from_existing",
"size": 17538,
"sha": ""
}
|
# informed_binn-sandbox.ipynb
Repository: loucerac/robustness
<code>
# %%
# https://www.sc-best-practices.org/conditions/gsea_pathway.html#id380
# Kang HM, Subramaniam M, Targ S, et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation
# Nat Biotechnol. 2020 Nov;38(11):1356]. Nat Biotechnol. 2018;36(1):89-94. doi:10.1038/nbt.4042
# %%
import sys
from pathlib import Path
import dotenv
import numpy as np
import pandas as pd
import scanpy as sc
from isrobust.bio import (
build_hipathia_renamers,
get_adj_matrices,
get_reactome_adj,
sync_gexp_adj,
)
from isrobust.datasets import load_kang
from isrobust.utils import set_all_seeds
from sklearn.model_selection import train_test_split
from sklearn.preprocessing import minmax_scale
model_kind = "binn-reactome"
debug = 1
seed = 42
model_kind = str(model_kind)
debug = bool(int(debug))
seed = int(seed)
project_path = Path(dotenv.find_dotenv()).parent
results_path = project_path.joinpath("results")
results_path.mkdir(exist_ok=True, parents=True)
data_path = project_path.joinpath("data")
data_path.mkdir(exist_ok=True, parents=True)
mygene_path = data_path.joinpath("mygene")
mygene_path.mkdir(exist_ok=True, parents=True)
figs_path = results_path.joinpath("figs")
figs_path.mkdir(exist_ok=True, parents=True)
tables_path = results_path.joinpath("tables")
tables_path.mkdir(exist_ok=True, parents=True)
set_all_seeds(seed=seed)
sc.set_figure_params(dpi=300, color_map="inferno")
sc.settings.verbosity = 1
sc.logging.print_header()
print(f"{debug=} {model_kind=}")
</code>
<code>
atlas = sc.read(
data_path.joinpath("theis", "pbmc_vars_sb.h5ad"),
cache=False,
)
atlas = atlas[atlas.obs['study']!='Villani'].copy()
atlas.X = atlas.layers["counts"].copy()
sc.pp.normalize_total(atlas)
sc.pp.log1p(atlas)
</code>
<code>
atlas.obs
</code>
<code>
adata.obs.cell_type
</code>
<code>
# %%
adata = load_kang(data_folder=data_path, normalize=True, n_genes=4000)
# %%
x_trans = adata.to_df()
obs = adata.obs
</code>
<code>
# %%
circuit_adj, circuit_to_pathway_adj = get_adj_matrices(
gene_list=x_trans.columns.to_list()
)
circuit_renamer, pathway_renamer, circuit_to_effector = build_hipathia_renamers()
kegg_circuit_names = circuit_adj.rename(columns=circuit_renamer).columns
kegg_pathway_names = circuit_to_pathway_adj.rename(columns=pathway_renamer).columns
circuit_adj.head()
# %%
reactome = get_reactome_adj()
reactome_pathway_names = reactome.columns
x_trans, circuit_adj = sync_gexp_adj(gexp=x_trans, adj=circuit_adj)
</code>
<code>
# %%
def train_val_test_split(features, val_size, test_size, stratify, seed):
train_size = 1 - (val_size + test_size)
x_train, x_test, y_train, y_test = train_test_split(
features,
stratify,
train_size=train_size,
stratify=stratify,
random_state=seed,
)
x_val, x_test, y_val, y_test = train_test_split(
x_test,
y_test,
test_size=test_size / (test_size + val_size),
stratify=y_test,
random_state=seed,
)
x_train = x_train.astype("float32")
x_val = x_val.astype("float32")
x_test = x_test.astype("float32")
return x_train, x_val, x_test, y_train, y_val, y_test
</code>
<code>
from sklearn.preprocessing import LabelEncoder,OneHotEncoder
x_train, x_val, x_test, y_train, y_val, y_test = train_val_test_split(
x_trans.apply(minmax_scale),
val_size=0.20,
test_size=0.20,
stratify=obs["cell_type"].astype(str) + obs["condition"].astype(str),
seed=42,
)
y_train = obs["cell_type"][y_train.index]
y_val = obs["cell_type"][y_val.index]
y_test = obs["cell_type"][y_test.index]
label_encoder = OneHotEncoder(sparse_output=False).fit(y_train.to_frame())
y_train_encoded = label_encoder.transform(y_train.to_frame())
y_val_encoded = label_encoder.transform(y_val.to_frame())
y_test_encoded = label_encoder.transform(y_test.to_frame())
</code>
<code>
from binn import BINN, Network
input_data = pd.read_csv("../../BINN/data/test_qm.csv", sep=",")
translation = pd.read_csv("../../BINN/data/translation.tsv", sep="\t")
pathways = pd.read_csv("../../BINN/data/pathways.tsv", sep="\t").rename(columns={"parent": "source", "child": "target"})
network = Network(
input_data=input_data,
pathways=pathways,
mapping=translation,
)
</code>
<code>
mygene_path.as_posix
</code>
<code>
TRANSLATE_GENES = True
if TRANSLATE_GENES:
import mygene
mg = mygene.MyGeneInfo()
mg.set_caching(mygene_path.as_posix())
gene_trans_df = mg.querymany(translation.input.unique(), scopes="uniprot", fields="symbol", as_dataframe=True, species='human', returnall=True)
gene_trans_df
</code>
<code>
translation = translation.merge(gene_trans_df["out"].dropna(subset="symbol").reset_index(names="input")[["input", "symbol"]],
how="left",
).dropna(subset="symbol").drop(["input", "Unnamed: 0"], axis=1).rename(columns={"symbol": "input"})
translation
</code>
<code>
genes = x_trans.columns.intersection(translation.input)
translation = translation.loc[translation.input.isin(genes)]
x_trans = x_trans.loc[:, genes]
</code>
<code>
kegg_trans = circuit_adj.melt(ignore_index=False).reset_index(names="symbol").query("value>0").rename(columns={"symbol": "source", "circuit": "target"}).drop("value", axis=1)
kegg_trans.head()
</code>
<code>
network = Network(
input_data=x_trans.T.reset_index(names="gene"),
pathways=pathways,
input_data_column="gene"
)
</code>
<code>
from sklearn.preprocessing import LabelEncoder,OneHotEncoder
x_train, x_val, x_test, y_train, y_val, y_test = train_val_test_split(
x_trans.apply(minmax_scale),
val_size=0.20,
test_size=0.20,
stratify=obs["cell_type"].astype(str) + obs["condition"].astype(str),
seed=42,
)
y_train = obs["cell_type"][y_train.index]
y_val = obs["cell_type"][y_val.index]
y_test = obs["cell_type"][y_test.index]
label_encoder = OneHotEncoder(sparse_output=False).fit(y_train.to_frame())
y_train_encoded = label_encoder.transform(y_train.to_frame())
y_val_encoded = label_encoder.transform(y_val.to_frame())
y_test_encoded = label_encoder.transform(y_test.to_frame())
</code>
<code>
batch_size = 32
callback = callbacks.EarlyStopping(
monitor="val_loss", # Stop training when `val_loss` is no longer improving
min_delta=1e-1, # "no longer improving" being defined as "no better than 1e-5 less"
patience=100, # "no longer improving" being further defined as "for at least 3 epochs"
verbose=0,
)
vae, encoder, decoder = build_kegg_vae(
circuits=circuit_adj, pathways=circuit_to_pathway_adj, seed=seed
)
history = vae.fit(
x_train.values,
shuffle=True,
verbose=0,
epochs=N_EPOCHS,
batch_size=batch_size,
callbacks=[callback],
validation_data=(x_val.values, None),
)
</code>
<code>
network = Network(
input_data=x_trans.T.reset_index(names="gene"),
pathways=pathways,
mapping=translation,
input_data_column="gene"
)
</code>
<code>
from binn import BINNClassifier, BINN
import torch
from torch.utils.data import DataLoader, TensorDataset
import lightning.pytorch as pl
device = torch.device("cuda")
binn = BINN(
n_layers=2,
network=network,
n_outputs=y_train_encoded.shape[1],
device="cuda"
)
binn
</code>
<code>
import os
n_workers = os.cpu_count() - 1
n_workers
</code>
<code>
dataloader = DataLoader(
dataset=TensorDataset(torch.Tensor(x_train.values), torch.Tensor(y_train_encoded)),
batch_size=32,
shuffle=True,
num_workers=n_workers
)
</code>
<code>
x_train.values.shape
</code>
<code>
trainer = pl.Trainer(
callbacks=[],
max_epochs=10,
)
trainer.fit(binn, dataloader)
</code>
<code>
def get_importances(data, abs=False):
if abs:
return np.abs(data).mean(axis=0)
v else:
return data.mean(axis=0)
def get_actiations(act_model, layer_id, data):
data_encoded = act_model.predict(data)[layer_id]
return data_encoded
# %%
def train_val_test_split(features, val_size, test_size, stratify, seed):
train_size = 1 - (val_size + test_size)
x_train, x_test, y_train, y_test = train_test_split(
features,
stratify,
train_size=train_size,
stratify=stratify,
random_state=seed,
)
x_val, x_test = train_test_split(
x_test,
test_size=test_size / (test_size + val_size),
stratify=y_test,
random_state=seed,
)
x_train = x_train.astype("float32")
x_val = x_val.astype("float32")
x_test = x_test.astype("float32")
return x_train, x_val, x_test
</code>
<code>
# %%
results_path_model = results_path.joinpath(model_kind)
obs = adata.obs.copy()
results_path_model.mkdir(exist_ok=True, parents=True)
</code>
<code>
# %%
x_train, x_val, x_test = train_val_test_split(
x_trans.apply(minmax_scale),
val_size=0.20,
test_size=0.20,
stratify=obs["cell_type"].astype(str) + obs["condition"].astype(str),
seed=42,
)
if model_kind == "ivae_kegg":
vae, encoder, decoder = build_kegg_vae(
circuits=circuit_adj, pathways=circuit_to_pathway_adj, seed=seed
)
elif model_kind == "ivae_reactome":
vae, encoder, decoder = build_reactome_vae(reactome, seed=seed)
else:
raise NotImplementedError("Model not yet implemented.")
batch_size = 32
callback = callbacks.EarlyStopping(
monitor="val_loss", # Stop training when `val_loss` is no longer improving
min_delta=1e-1, # "no longer improving" being defined as "no better than 1e-5 less"
patience=100, # "no longer improving" being further defined as "for at least 3 epochs"
verbose=0,
)
history = vae.fit(
x_train.values,
shuffle=True,
verbose=0,
epochs=N_EPOCHS,
batch_size=batch_size,
callbacks=[callback],
validation_data=(x_val.values, None),
)
evaluation = {}
evaluation["train"] = vae.evaluate(
x_train, vae.predict(x_train), verbose=0, return_dict=True
)
evaluation["val"] = vae.evaluate(x_val, vae.predict(x_val), verbose=0, return_dict=True)
evaluation["test"] = vae.evaluate(
x_test, vae.predict(x_test), verbose=0, return_dict=True
)
pd.DataFrame.from_dict(evaluation).reset_index(names="metric").assign(seed=seed).melt(
id_vars=["seed", "metric"],
value_vars=["train", "val", "test"],
var_name="split",
value_name="score",
).assign(model=model_kind).to_pickle(
results_path_model.joinpath(f"metrics-seed-{seed:02d}.pkl")
)
layer_outputs = [layer.output for layer in encoder.layers]
activation_model = Model(inputs=encoder.input, outputs=layer_outputs)
# only analyze informed and funnel layers
for layer_id in range(1, len(layer_outputs)):
if model_kind == "ivae_kegg":
if layer_id == 1:
colnames = kegg_circuit_names
layer_name = "circuits"
elif layer_id == 2:
colnames = kegg_pathway_names
layer_name = "pathways"
elif layer_id == (len(layer_outputs) - 1):
n_latents = len(kegg_pathway_names) // 2
colnames = [f"latent_{i:02d}" for i in range(n_latents)]
layer_name = "funnel"
else:
continue
elif model_kind == "ivae_reactome":
if layer_id == 1:
colnames = reactome_pathway_names
layer_name = "pathways"
elif layer_id == (len(layer_outputs) - 1):
n_latents = len(reactome_pathway_names) // 2
colnames = [f"latent_{i:02d}" for i in range(n_latents)]
layer_name == "funnel"
else:
continue
else:
raise NotImplementedError("Model not yet implemented.")
print(f"encoding layer {layer_id}")
encodings = get_activations(
act_model=activation_model,
layer_id=layer_id,
data=x_trans.apply(minmax_scale),
)
encodings = pd.DataFrame(encodings, index=x_trans.index, columns=colnames)
encodings["split"] = "train"
encodings.loc[x_val.index, "split"] = "val"
encodings.loc[x_test.index, "split"] = "test"
encodings["layer"] = layer_name
encodings["seed"] = seed
encodings["model"] = model_kind
encodings = encodings.merge(
obs[["cell_type", "condition"]],
how="left",
left_index=True,
right_index=True,
)
encodings.to_pickle(
results_path_model.joinpath(
f"encodings_layer-{layer_id:02d}_seed-{seed:02d}.pkl"
)
)
</code>
|
{
"filename": "informed_binn-sandbox.ipynb",
"repository": "loucerac/robustness",
"query": "transformed_from_existing",
"size": 91206,
"sha": ""
}
|
# notes_gan_1.ipynb
Repository: xiptos/is
[](https://colab.research.google.com/github/xiptos/is_notes/blob/main/gan.ipynb)
# Generative Adversarial Networks (GAN)
Are based on a strategy where two different deep networks are pitted against one another, with the goal of getting **one network to create new samples** that are not from the training data, but are so much like the training data that the other **network can’t tell the difference**.
In this example, let's train a generator to produce gaussian distribution values based on random noise.
## Import libraries
<code>
import matplotlib.pyplot as plt
#importing Libraries
import numpy as np
import torch
import torch.nn as nn
</code>
## Understanding data
<code>
from sklearn.datasets import make_blobs
from scipy.stats import multivariate_normal
X1, Y1 = make_blobs(n_samples=500, centers=[(5, 5)], n_features=3, random_state=0)
fig = plt.figure(figsize=(18, 8))
#Parameters to set
mu_x = 5
variance_x = 0.5
mu_y = 5
variance_y = 0.5
#Create grid and multivariate normal
x = np.linspace(3, 7, 20)
y = np.linspace(3, 7, 20)
X, Y = np.meshgrid(x, y)
pos = np.empty(X.shape + (2,))
pos[:, :, 0] = X;
pos[:, :, 1] = Y
rv = multivariate_normal([mu_x, mu_y], [[variance_x, 0], [0, variance_y]])
ax1 = fig.add_subplot(1, 2, 1, projection='3d')
surf = ax1.plot_surface(X, Y, rv.pdf(pos) * 3, rstride=1, cstride=1, linewidth=1, antialiased=False, cmap='viridis')
ax2 = fig.add_subplot(1, 2, 2)
ax2.scatter(X1[:, 0], X1[:, 1], marker="o", c=Y1, s=25, edgecolor="k")
cc = plt.Circle((5, 5), 1, fill=False, edgecolor='red', linewidth=2)
ax2.set_aspect(1)
ax2.add_artist(cc)
plt.show()
</code>
<code>
print(X1.shape)
print(Y1.shape)
</code>
<code>
def generate_norm_data(batch_size: int = 16):
X1, Y1 = make_blobs(n_samples=batch_size, centers=[(5, 5)], n_features=3)
return X1
</code>
<code>
from sklearn.datasets import make_blobs
X1 = generate_norm_data(1000)
plt.figure(figsize=(8, 8))
ax2 = plt.gca()
plt.title("One informative feature, one cluster per class", fontsize="small")
ax2.scatter(X1[:, 0], X1[:, 1], marker="o", s=25, edgecolor="k")
cc = plt.Circle((5, 5), 1, fill=False, edgecolor='red', linewidth=2)
ax2.set_aspect(1)
ax2.add_artist(cc)
plt.show()
</code>
<code>
from torch.utils.data import Dataset
from torch.utils.data import DataLoader
class NormDataset(Dataset):
def __init__(self, n_samples=1000):
self.Xs, self.y = make_blobs(n_samples=n_samples, centers=[(5, 5)], n_features=3)
def __len__(self):
return len(self.Xs)
def __getitem__(self, idx):
image = self.Xs[idx].astype(np.float32)
label = self.y[idx]
return image, label
</code>
## The generator
<code>
class Generator(nn.Module):
def __init__(self):
super().__init__()
self.g = torch.nn.Sequential(
torch.nn.Linear(4, 16),
torch.nn.BatchNorm1d(16),
torch.nn.LeakyReLU(),
torch.nn.Linear(16, 2)
)
def forward(self, x):
return self.g(x)
</code>
## The discriminator
<code>
class Discriminator(nn.Module):
def __init__(self):
super().__init__()
self.d = torch.nn.Sequential(
torch.nn.Linear(2, 64),
torch.nn.LeakyReLU(),
torch.nn.Linear(64, 64),
torch.nn.LeakyReLU(),
torch.nn.Linear(64, 1),
torch.nn.Sigmoid()
)
def forward(self, x):
res = self.d(x)
return res
</code>
## Training
<code>
if torch.cuda.is_available():
device = torch.device("cuda")
elif torch.backends.mps.is_available():
device = torch.device("mps")
else:
device = torch.device("cpu")
print(f"Using device: {device}")
</code>
<code>
epochs = 1000
batch_size = 1000
training_data = NormDataset(n_samples=10000)
dataloader = DataLoader(training_data, batch_size=batch_size)
G = Generator()
D = Discriminator()
D.to(device)
G.to(device)
# Optimizers
G_optimizer = torch.optim.Adam(G.parameters(), lr=0.0002)
D_optimizer = torch.optim.Adam(D.parameters(), lr=0.0002)
loss = nn.BCELoss()
D_losses = []
G_losses = []
test_data = []
for epoch in range(epochs):
for idx, (true_data, _) in enumerate(dataloader):
# Training the discriminator
# Real inputs are actual examples with gaussian distribution
# Fake inputs are from the generator
# Real inputs should be classified as 1 and fake as 0
real_inputs = true_data.to(device)
real_outputs = D(real_inputs)
real_label = torch.ones(real_inputs.shape[0], 1).to(device)
noise = torch.tensor(np.random.normal(0, 1, (real_inputs.shape[0], 4))).float()
noise = noise.to(device)
fake_inputs = G(noise)
fake_outputs = D(fake_inputs)
fake_label = torch.zeros(fake_inputs.shape[0], 1).to(device)
outputs = torch.cat((real_outputs, fake_outputs), 0)
targets = torch.cat((real_label, fake_label), 0)
D_loss = loss(outputs, targets)
D_optimizer.zero_grad()
D_loss.backward()
D_optimizer.step()
# Training the generator
# For generator, goal is to make the discriminator believe everything is 1
noise = torch.tensor(np.random.normal(0, 1, (real_inputs.shape[0], 4))).float()
noise = noise.to(device)
fake_inputs = G(noise)
fake_outputs = D(fake_inputs)
fake_targets = torch.ones([fake_inputs.shape[0], 1]).to(device)
G_loss = loss(fake_outputs, fake_targets)
G_optimizer.zero_grad()
G_loss.backward()
G_optimizer.step()
G_losses.append(G_loss.item())
D_losses.append(D_loss.item())
if (epoch + 1) % 100 == 0:
print('Epoch {} Iteration {}: discriminator_loss {:.3f} generator_loss {:.3f}'.format(epoch, idx, D_loss.item(), G_loss.item()))
test = (torch.rand(real_inputs.shape[0], 4) - 0.5) / 0.5
noise = torch.tensor(np.random.normal(0, 1, (real_inputs.shape[0], 4))).float().to(device)
test_data.append(G(noise).detach().cpu().numpy())
</code>
## Check training
<code>
print(len(test_data[0]))
</code>
<code>
#plot the loss function
plt.plot(range(len(D_losses)), D_losses)
plt.plot(range(len(G_losses)), G_losses)
plt.ylabel('Loss')
plt.ylabel('batches')
</code>
## Evolution of the GAN training
<code>
#noise = torch.randn(size=(500, 4)).cuda()
#noise = (torch.rand(real_inputs.shape[0], 4) - 0.5) / 0.5
#noise = torch.tensor(np.random.normal(0, 1, (500, 4))).float().cuda()
#print(noise.shape)
fig = plt.figure(figsize=(18, 30))
#generated_data = G(noise).detach().cpu().numpy()
for i, generated_data in enumerate(test_data):
plt.subplot(8, 4, i + 1)
ax2 = plt.gca()
plt.title("Epoc %d" % i, fontsize="small")
ax2.scatter(generated_data[:, 0], generated_data[:, 1], marker="o", s=25, edgecolor="k")
cc = plt.Circle((5, 5), 1, fill=False, edgecolor='red', linewidth=2)
ax2.set_aspect(1)
ax2.add_artist(cc)
plt.show()
</code>
|
{
"filename": "notes_gan_1.ipynb",
"repository": "xiptos/is",
"query": "transformed_from_existing",
"size": 14252,
"sha": ""
}
|
# 2.SOG_3.ipynb
Repository: xiaojierzi/iSORT
<code>
from iSORTlib import *
seed_everything(20)
</code>
# Tutorial for finding spatial-organzing genes (SOGs) and performing in silico knockouts
## 1. Load data
### Set directories
<code>
sc_data_dir = '151674_data.csv'
sc_meta_dir = '151674_meta.csv'
st_data_dir = '151675_data.csv'
st_meta_dir = '151675_meta.csv'
</code>
## 2. Data preprocessing
### Read and preprocess scRNA-seq and ST data
<code>
sc_adata = preprocess.read_data(sc_data_dir, sc_meta_dir, 'sc')
st_adata = preprocess.read_data(st_data_dir, st_meta_dir, 'st')
</code>
### Identify and align common highly variable genes
<code>
common_hvg_data = preprocess.get_common_hvg(sc_adata, st_adata)
sc_adata, st_adata = common_hvg_data
</code>
### Normalize spatial coordinates
<code>
spatial = normalize_spatial_coordinates(st_adata)
</code>
## 3. Calculate weights
<code>
kliep = KLIEP(sigma=22)
kliep.fit(sc_adata)
weights_st = kliep.calculate_weights(st_adata)
</code>
## 4. Training
### Initialize and train model with spatial data and weights (Remember changing the number of input_feautres)
<code>
model, criterion, optimizer = initialize_model(input_features=1317, hidden_layers=[324, 128, 64], output_features=2)
train_loader, val_loader=prepare_data(st_adata.X, spatial, weights_st)
train_and_validate(model, train_loader, val_loader, optimizer, criterion, 300)
</code>
## 5. Generate predictions and visualizing
<code>
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
test=torch.tensor(sc_adata.X, dtype=torch.float32)
with torch.no_grad():
test=test.to(device)
predictions_normalized = model(test)
predictions_normalized=predictions_normalized.to('cpu')
predictions_array = predictions_normalized.numpy()
sc_adata.obsm['prediction_space'] = predictions_array
plt.rcParams["figure.figsize"] = (3, 3)
sc.pl.embedding(sc_adata, basis='prediction_space', color='Layer_Guess')
</code>
## 6. Identify SOGs
<code>
top_50_gene_indices, top_50_gene_names = SOGlocate.compute_important_genes_and_indices(model, sc_adata, 50, 2)
</code>
## 7. Perform in silico knockouts
<code>
SOGlocate.knockout_and_visualize(model, test, top_50_gene_indices, sc_adata, 'Layer_Guess', 'Top 50 Genes Knockout')
</code>
|
{
"filename": "2.SOG_3.ipynb",
"repository": "xiaojierzi/iSORT",
"query": "transformed_from_existing",
"size": 217537,
"sha": ""
}
|
# plotly_2.ipynb
Repository: siobhon-egan/internship
# Visualizing bioinformatics data with plot.ly
This notebook is used for visualising the quality scores of each sample.
It generates an interactive graph, one per sample of all the seqeuences generated from that sample.
https://plot.ly/~johnchase/22/visualizing-bioinformatics-data-with-plo/
<code>
!conda search colorlover
</code>
<code>
# Obtain files
</code>
<code>
!wget https://github.com/johnchase/plotly-notebook/raw/master/raw_data.tar.gz
</code>
<code>
!tar -xvzf raw_data.tar.gz
</code>
<code>
#load libraries
</code>
<code>
import plotly.plotly as py
import plotly.graph_objs as go
from plotly.tools import FigureFactory as FF
import colorlover as cl
import skbio
from skbio.alignment import global_pairwise_align_nucleotide
from skbio.sequence import DNA
import pandas as pd
import itertools
import numpy as np
</code>
<code>
import plotly
plotly.tools.set_credentials_file(username='siobhonegan', api_key='4RpshP5pf7nc7tyGvhaD')
</code>
<code>
py.sign_in('siobhonegan', '')
</code>
## 1. Sequence Quality
Because the quality of sequence data produced by high throughput sequencing varies between sequencing runs and samples it is important to look at the sequence quality and possibly filter or trim sequences, or remove samples where the quality is low. A more detailed description of the fastq format and quality scores can be found here [http://scikit-bio.org/docs/latest/generated/skbio.io.format.fastq.html?highlight=fastq#module-skbio.io.format.fastq]. Quality scores themselves are difficult to interpret, so we will use scikit-bio to decode the scores for us. Here we load the sequence data into a generator with scikit-bio.
<code>
f = '319ITF-B16_S26_L001_R1_001.fastq'
seqs = skbio.io.read(f, format='fastq', verify=False, variant='illumina1.8')
</code>
We can view one of the skbio.sequence._sequence.Sequence entries in the generator object. This will display the sequence data and associated metadata.
<code>
seq1 = seqs.__next__()
seq1
</code>
Looking through sequence quality on a per-sequence basis is tedious and it would be diffult to decipher meaningful patterns, plotting the data is a better solution. In order to create a meaningful plot we will first create a pd.DataFrame object of the quality scores. Due to limitations in data size and because a subset of our data will fairly accurately represent the quality of the full data set we will only look at the first 500 sequences in our data set.
<code>
seqs = skbio.io.read(f, format='fastq', verify=False, variant='illumina1.8')
df = pd.DataFrame()
num_sequences = 500
for count, seq in enumerate(itertools.islice(seqs, num_sequences)):
df[count] = seq.positional_metadata.quality
</code>
Now that we have a dataframe with all of our quality scores it is easy to visualize them with plotly.
We can improve upon a basic boxplot by defining a specific color scheme. Fastq quality scores range from 0-40, and poor quality is often considered to be anything less then 20. Given this we will define a diverging colormap where an average quality score of below 20 will be a shade of red and anything above will be a shade of blue. (20 will be yellow). This will help with distinguishing regions of high versus low quality.
First define the colormap using color lover
<code>
purd = cl.scales['11']['div']['RdYlBu']
purd40 = cl.interp(purd, 40)
</code>
Now we can make boxplots of the quality scores on a per base basis
<code>
traces = []
for e in range(len(df)):
traces.append(go.Box(
y=df.iloc[e].values,
name=e,
boxpoints='none',
whiskerwidth=0.2,
marker=dict(
size=.1,
color=purd40[int(round(df.iloc[e].mean(), 0))]
),
line=dict(width=1),
))
layout = go.Layout(
title='Quality Score Distributions',
yaxis=dict(
title='Quality Score',
autorange=True,
showgrid=True,
zeroline=True,
gridcolor='#d9d4d3',
zerolinecolor='#d9d4d3',
),
xaxis=dict(
title='Base Position',
),
font=dict(family='Times New Roman', size=16, color='#2e1c18'),
paper_bgcolor='#eCe9e9',
plot_bgcolor='#eCe9e9'
)
fig = go.Figure(data=traces, layout=layout)
py.iplot(fig, filename='quality-scores')
</code>
## 2. Sequence Alignment
One of the most important steps in bioinformatics is sequence alignment. Aligning sequences helps us to understand the relationship between two or more sequences, it allows us to identify specific bases or regions that vary between sequences and can be used to discover novel sequences. For the purpose of this notebook we will use the global pariwise aligner from skbio this is known to be slow and should be updated soon. If you are aligning more than a few sequences, there are other faster aligners that would be preferrable such as MAFFT. QIIME2 provides a convenient set of tools that wrap aligners such as MAFFT.
### Align the first two sequences using scikit-bio
This is slow, and really only appropriate for educational purposes. In fact scikit-bio will generate a warning to this effect. You can find more information about the scikit-bio aligner in this github issue. If you are aligning sequences locally scikit-bio has optimized algorithms appropriate for larger scale data.
Once again we load the sequences using scikit-bio. Here we will only load the first two sequences in order to illustrate pairwise alignment.
<code>
seqs = [DNA(e) for e in itertools.islice(skbio.io.read(f, format='fastq', variant='illumina1.8'), 2)]
</code>
<code>
aligned_seqs = global_pairwise_align_nucleotide(seqs[0], seqs[1])
aligned_seqs
</code>
### Align multiple sequences
Pairwise alignment is a more simple task than multiple sequence alignment. With multiple sequences each additional sequence changes the overall alignment, meaning it must be constructed progressively with each additional sequence. This can be very computationally expensive. If you do not wish to wait for the alignment to run (or do not wish to install An Introduction to Bioinformatics, you can leave the following lines commented out and load the pre-aligned sequences.
<code>
# from iab.algorithms import progressive_msa, tree_from_distance_matrix
# from functools import partial
# f = 'run1_16s/rev_seqs/1AM1JR7QWMSFA.fastq'
# seqs = [DNA(e) for e in itertools.islice(skbio.io.read(f, format='fastq', verify=False, variant='illumina1.8'), 10)]
# seqs = [e for e in seqs if not e.has_degenerates()]
# msa = progressive_msa(seqs, global_pairwise_align_nucleotide)
# msa.write('msa10.fna')
msa = skbio.alignment.TabularMSA.read('msa10.fna', constructor=DNA)
</code>
In order to create a meaningful visualization of the multiple sequence alignment we will use heatmap functionality of plotly.
First we assign a numeric value to each possible character in our alignment, "A", "T", "G", "C", and "-"
<code>
base_dic = {'A': 1, 'C': .25, 'G': .5, 'T': .75, '-': 0}
</code>
Next we define a function that takes an alignment and returns two, two-dimensional arrays, one of the characters in the alignment and one of the numeric value that represents the character defined in the dictionary above. The numeric value will be used to define the color in the heatmap. The function below will create an array of the alignment such the only bases that are colored differently are bases that are different from the first sequence. This will make identifying differences in sequences much easier. If each based was given a unique color regardless of it's relationship to other sequences the plot would be noisy and difficult to interpret.
<code>
def seq_align_for_plot(msa):
base_text = [list(str(e)) for e in msa]
base_values = np.zeros((len(base_text), len(base_text[0])))
for i in range(len(base_text[0])):
for j in range(len(base_text)):
if base_text[j][i] != base_text[0][i]:
base_values[j][i] = base_dic[base_text[j][i]]
return(base_text, base_values)
base_text, base_values = seq_align_for_plot(msa)
</code>
Define a colorscale where the values for each base is given a defined color
<code>
colorscale=[[0.00, '#F4F0E4'],
[0.25, '#1b9e77'],
[0.50, '#d95f02'],
[0.75, '#7570b3'],
[1.00, '#e7298a']]
</code>
Create a list of arbitrary sequences names (The original sequence names were randomly generated and do not have meaning associated with them).
<code>
seq_names = ["Seq " + str(e + 1) for e in range(len(base_text))]
</code>
Finally we can plot the alignment.
<code>
fig = FF.create_annotated_heatmap(base_values,
annotation_text=base_text,
colorscale=colorscale)
fig['layout'].update(
title="Aligned Sequences",
xaxis=dict(ticks='',
side='top',
ticktext=list(np.arange(0, len(base_text[0]), 10)),
tickvals=list(np.arange(0, len(base_text[0]), 10)),
showticklabels=True,
tickfont=dict(family='Bookman',
size=18,
color='#22293B',
),
),
yaxis=dict(autorange='reversed',
ticks='',
ticksuffix=' ',
ticktext=seq_names,
tickvals=list(np.arange(0, len(base_text))),
showticklabels=True,
tickfont=dict(family='Bookman',
size=18,
color='22293B',
),
),
width=10000,
height=450,
autosize=True,
annotations=dict(font=dict(family='Courier New, monospace',
size=14,
color='#3f566d'
),
)
)
py.iplot(fig, filename='msa')
</code>
|
{
"filename": "plotly_2.ipynb",
"repository": "siobhon-egan/internship",
"query": "transformed_from_existing",
"size": 24217,
"sha": ""
}
|
# Exploration_1.ipynb
Repository: krassowski/multi-omics-state-of-the-field
**Aims**:
- list high-impact works to aid navigation of the field
- check for unexpectedly common authors/affiliations/journals to screening for potential false-positive matches (see the Integromics and Panomics companies)
<code>
%run notebook_setup.ipynb
</code>
<code>
%vault from pubmed_derived_data import literature, affiliations, authors, publication_types
</code>
<code>
%vault from journals_data import web_of_science_journals, scimago_by_issn
</code>
<code>
literature['journal_sjr_rank'] = (
literature['journal_issn']
.str.replace('-', '')
.fillna('-')
.apply(
lambda x: (
scimago_by_issn.loc[x].Rank
if x in scimago_by_issn.index else
None
)
)
)
</code>
## A quick overview/hot-takes
<code>
columns_to_show = ['title', 'journal', 'doi', 'journal_sjr_rank']
</code>
<code>
def display_sorted(data):
return data[columns_to_show].sort_values(['journal_sjr_rank', 'title'])
</code>
### Benchmarks
<code>
literature[literature.title.str.lower().str.contains('|'.join(['benchmark', 'evaluation', 'comparison']))].pipe(display_sorted)
</code>
### Biomarkers
<code>
literature[literature.title.str.lower().str.contains('|'.join(['biomarker']))].pipe(display_sorted)
</code>
<code>
literature.pipe(display_sorted).head(20)
</code>
### Discoveries?
<code>
literature[literature.title.str.lower().str.contains('|'.join(['reveals']))].pipe(display_sorted).head(20)
</code>
### Affiliations
Most authors with given affiliation on papers:
<code>
affiliations.Affiliation.sorted_value_counts().head(5).to_frame()
</code>
Most papers with given affiliation:
<code>
affiliations[['Affiliation', 'PMID']].drop_duplicates().Affiliation.sorted_value_counts().head(10).to_frame()
</code>
We were previously getting false hits because we were matching by affiliations:
- "Multi-Omics Based Creative Drug Research Team, Kyungpook National University, Daegu 41566, Republic of Korea"
- "Panomics, Inc"
so it is important to check if no affiliations overrepresented. Would need a cleanup to be more reliable (not a priority).
### Authors
Note: not neccessarily unique persons, adoption of ORCID still low:
<code>
authors['JointName'].sorted_value_counts().head(15).to_frame()
</code>
### Publication kind and type
<code>
literature.kind.sorted_value_counts()
</code>
<code>
sum(literature['Is Review'] == True)
</code>
<code>
publication_types['0'].sorted_value_counts().to_frame('count')
</code>
<code>
literature[literature['Is English Abstract'] == True][['title', 'doi']]
</code>
<code>
literature[literature['Is Interview'] == True][['title', 'doi']]
</code>
<code>
literature[literature['Is Congress'] == True][['title', 'doi']]
</code>
<code>
literature[literature['Is News'] == True][['title', 'doi']]
</code>
<code>
literature[literature['Is Dataset'] == True][['title', 'doi']]
</code>
### Journals
<code>
journal_freq = literature.journal.sorted_value_counts()
</code>
<code>
journal_freq.head(20).to_frame()
</code>
Sanity check (is any of the top names not unique?) - the numbers should be same if counting by ISSN:
<code>
literature.journal_issn.sorted_value_counts().head(10)
</code>
<code>
journal_freq[journal_freq < 50]
</code>
<code>
literature = literature.replace({float('nan'): None}).infer_objects()
%R -i literature
</code>
## Publication types
<code>
%%R
library(ComplexUpset)
source('helpers/plots.R')
source('helpers/colors.R')
</code>
<code>
publication_types_list = ['Is ' + t for t in publication_types['0'].sorted_value_counts().where(lambda x: x > 10).dropna().index]
</code>
<code>
%%R -i publication_types_list -w 2000 -r 100 -h 800
upset(
literature,
publication_types_list,
base_annotations=list(
'Intersection size'=intersection_size(
text=list(angle=90, vjust=0.5, hjust=0)
)
),
width_ratio=0.1,
set_sizes=(
upset_set_size(
geom=geom_bar(width=0.5)
)
+ scale_y_continuous(trans=reverse_log_trans())
+ theme(axis.text.x=element_text(angle=90))
)
)
</code>
<code>
%%R -w 700 -h 400 -r 100
(
ggplot(literature, aes(x=year, fill=has_doi))
+ geom_bar()
+ theme_bw()
) + (
ggplot(literature, aes(x=year, fill=has_pmc))
+ geom_bar()
+ theme_bw()
) & plot_layout(ncol=1)
</code>
|
{
"filename": "Exploration_1.ipynb",
"repository": "krassowski/multi-omics-state-of-the-field",
"query": "transformed_from_existing",
"size": 284352,
"sha": ""
}
|
# basic_usage_2.ipynb
Repository: estorrs/enrichrpy
<code>
import enrichrpy.enrichr as een
import enrichrpy.plotting as epl
</code>
define some test genes
<code>
genes = [
'TYROBP',
'HLA-DRA',
'SPP1',
'LAPTM5',
'C1QB',
'FCER1G',
'GPNMB',
'FCGR3A',
'RGS1',
'HLA-DPA1',
'ITGB2',
'C1QC',
'HLA-DPB1',
'IFI30',
'SRGN',
'APOC1',
'CD68',
'HLA-DRB1',
'C1QA',
'LYZ',
'APOE',
'HLA-DQB1',
'CTSB',
'HLA-DQA1',
'CD74',
'AIF1',
'FCGR2A',
'CD14',
'S100A9',
'CTSS'
]
</code>
default gene set library is 'GO_Biological_Process_2021'
all possible gene set libraries are listed below
<code>
sorted(een.get_enrichr_libraries(names_only=True))
</code>
Querying genes against the GO_Biological_Process_2021 gene set library
<code>
df = een.get_pathway_enrichment(genes, gene_set_library='GO_Biological_Process_2021')
df
</code>
There are two main GSEA plots available, bar and dot
<code>
epl.enrichment_barplot(df, n=20)
</code>
<code>
epl.enrichment_dotplot(df, n=20, hue='Z-score', log=True)
</code>
|
{
"filename": "basic_usage_2.ipynb",
"repository": "estorrs/enrichrpy",
"query": "transformed_from_existing",
"size": 55112,
"sha": ""
}
|
# Prediction_Churn_Analysis_R.ipynb
Repository: RajEshwariCodes/Churn
<code>
import pandas as pd
import numpy as np
</code>
<code>
df=pd.read_csv("/content/drive/MyDrive/churn.csv")
</code>
<code>
df.head()
</code>
<code>
df.drop("customer_id",axis=1,inplace=True)
</code>
<code>
df
</code>
<code>
df.info() #structure of dataframe
</code>
<code>
from sklearn.preprocessing import LabelEncoder
label_encoder = LabelEncoder()
df['country'] = label_encoder.fit_transform(df['country'])
df['gender'] = label_encoder.fit_transform(df['gender'])
df
</code>
<code>
df.info()
</code>
<code>
from sklearn.preprocessing import MinMaxScaler
scaler = MinMaxScaler()
df_normalized = scaler.fit_transform(df)
</code>
<code>
df_normalized
</code>
<code>
X = df.drop('churn', axis=1)
y = df['churn']
</code>
<code>
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
</code>
<code>
from sklearn.tree import DecisionTreeClassifier
from sklearn.model_selection import train_test_split
from sklearn.metrics import accuracy_score
# Assuming X_train, X_test, y_train, y_test are already defined
# using train_test_split as explained in the previous responses
# Create a Decision Tree model
dt_classifier = DecisionTreeClassifier(random_state=42)
# Train the model
dt_classifier.fit(X_train, y_train)
# Make predictions on the test set
y_pred_dt = dt_classifier.predict(X_test)
# Evaluate the model
accuracy = accuracy_score(y_test, y_pred_dt)
print(f"Decision Tree Accuracy: {accuracy}")
</code>
<code>
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import train_test_split
from sklearn.metrics import accuracy_score
# Assuming X_train, X_test, y_train, y_test are already defined
# Create a Random Forest model
rf_classifier = RandomForestClassifier(random_state=42)
# Train the model
rf_classifier.fit(X_train, y_train)
# Make predictions on the test set
y_pred_rf = rf_classifier.predict(X_test)
# Evaluate the model
accuracy = accuracy_score(y_test, y_pred_rf)
print(f"Random Forest Accuracy: {accuracy}")
</code>
<code>
from sklearn.svm import SVC
from sklearn.model_selection import train_test_split
from sklearn.metrics import accuracy_score
from sklearn.preprocessing import StandardScaler
# Assuming X_train, X_test, y_train, y_test are already defined
# Standardize the features (scaling is often important for SVM)
scaler = StandardScaler()
X_train_scaled = scaler.fit_transform(X_train)
X_test_scaled = scaler.transform(X_test)
# Create an SVM model
svm_classifier = SVC(random_state=42)
# Train the model
svm_classifier.fit(X_train_scaled, y_train)
# Make predictions on the scaled test set
y_pred_svm = svm_classifier.predict(X_test_scaled)
# Evaluate the model
accuracy = accuracy_score(y_test, y_pred_svm)
print(f"SVM Accuracy: {accuracy}")
</code>
<code>
from sklearn.metrics import confusion_matrix
l1=[y_pred_svm,y_pred_rf,y_pred_dt]
for i in l1:
conf_matrix = confusion_matrix(y_test, i)
print("Confusion Matrix:")
print(conf_matrix)
</code>
<code>
from sklearn.metrics import precision_score, recall_score, f1_score
for y_pred in l1:
precision = precision_score(y_test, y_pred)
recall = recall_score(y_test, y_pred)
f1 = f1_score(y_test, y_pred)
print(f"Precision: {precision}")
print(f"Recall: {recall}")
print(f"F1 Score: {f1}")
</code>
<code>
from sklearn.metrics import classification_report
for y_pred in l1:
report = classification_report(y_test, y_pred)
print("Classification Report:")
print(report)
</code>
<code>
from sklearn.metrics import roc_curve, roc_auc_score
import matplotlib.pyplot as plt
for predicted_probabilities in l1:
# Assuming binary classification
fpr, tpr, thresholds = roc_curve(y_test, predicted_probabilities)
roc_auc = roc_auc_score(y_test, predicted_probabilities)
plt.plot(fpr, tpr, label=f'AUC = {roc_auc}')
plt.xlabel('False Positive Rate')
plt.ylabel('True Positive Rate')
plt.legend()
plt.show()
</code>
<code>
'''
H.W (any 2 hw on/before saturday)
reason behind every step and descions made, explain steps.
implement : min max scaling , report outcomes
implement : try implement other algorithms (naive bayes, knn etc).
'''
</code>
<code>
from sklearn.model_selection import train_test_split
from sklearn.naive_bayes import GaussianNB
from sklearn.metrics import accuracy_score
# Assuming you have a dataset X (features) and y (labels)
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Create a Naive Bayes classifier
nb_classifier = GaussianNB()
# Train the classifier
nb_classifier.fit(X_train, y_train)
# Make predictions
y_pred_nb = nb_classifier.predict(X_test)
# Evaluate the accuracy
accuracy = accuracy_score(y_test, y_pred_nb)
print(f'Accuracy: {accuracy}')
</code>
<code>
from sklearn.model_selection import train_test_split
from sklearn.neighbors import KNeighborsClassifier
from sklearn.metrics import accuracy_score
# Assuming you have a dataset X (features) and y (labels)
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Create a KNN classifier (let's say, k=3)
knn_classifier = KNeighborsClassifier(n_neighbors=3)
# Train the classifier
knn_classifier.fit(X_train, y_train)
# Make predictions
y_pred_knn = knn_classifier.predict(X_test)
# Evaluate the accuracy
accuracy = accuracy_score(y_test, y_pred_knn)
print(f'Accuracy: {accuracy}')
</code>
<code>
from sklearn.preprocessing import MinMaxScaler
# Create a MinMaxScaler object
scaler = MinMaxScaler()
# Fit the scaler on your data and transform it
scaled_data = scaler.fit_transform(df)
# The scaled_data variable now contains the MinMax-scaled values
print("Original Data:\n", df)
print("\nScaled Data:\n", scaled_data)
</code>
<code>
scaled_data
</code>
<code>
X = scaled_data.drop('churn', axis=1)
y = scaled_data['churn']
</code>
|
{
"filename": "Prediction_Churn_Analysis_R.ipynb",
"repository": "RajEshwariCodes/Churn",
"query": "transformed_from_existing",
"size": 184524,
"sha": ""
}
|
# wright_fisher-hints_1.ipynb
Repository: sparkingdark/dataset-test
This is an ipython notebook. Lectures about Python, useful both for beginners and experts, can be found at http://scipy-lectures.github.io.
I recommend installing the [Anaconda](https://store.continuum.io/cshop/academicanaconda) distribution. Make sure not to pay for it! Click Anaconda Academic License; it should be free for those with academic e-mail addresses.
Open the notebook by (1) copying this file into a directory, (2) in that directory typing
ipython notebook
and (3) selecting the notebook.
In this exercise, we will build a Wright-Fisher simulation model, which will be the basis of most of our simulation efforts.
# Wright-Fisher model
## Motivation
Population genetics seeks to describe and understand patterns of genetic diversity found in natural and artificial populations. Blood groups, which are genetically determined, have very uneven geographical distributions:
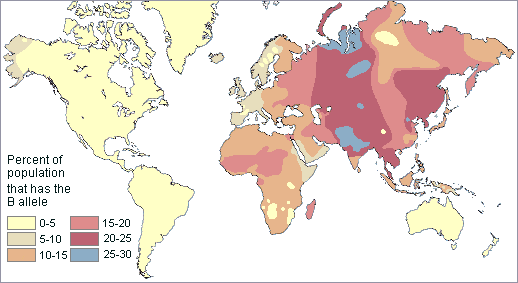
We can ask many questions about these distributions: Are these the result of natural selection? Of dramatic historical events? Or are they simply meaningless random distributions? To understand the data, we need to build models for the data that make predictions and can be falsified.
In this exercise, we will build a simple model for the evolution of allele frequencies in a population. We will use this model over and over in this course. To build an evolutionary model, we need to specify a number of parameters: the population size, the structure of the genome (e.g., the number of chromosomes, its ploidy), the mode of reproduction of the population (e.g., sexual vs asexual), whether individuals have mate choice preferences, the distribution in the number and timing of offspring by individual, how mutations arise, how alleles are transmitted from parent to offspring. We also have to decide on the starting point in our model, the ancestral population from which we will consider evolution. This represents a lot of choices. Our first model will try to keep everything as simple as possible:
* Population size: constant, $N$
* Structure of the genome: one single haploid chromosome of length 1 base pair.
* Mode of reproduction: asexual
* Mutation process: No mutation
* Transmission: Asexual transmission (clonal reproduction)
* Distribution of offspring: ?
It was straightforrward to come up with the simplest parameters so far, but here we need to think a bit more. Since we have a haploid population of constant size, the average number of offspring per individual must be one. If every individual has exactly one offspring, the population will never change, and the model will fail to account for the observed variation in reproductive success across individuals.
If we let each parent independently spawn a random number of offspring, the population size could change a little bit at every generation. That's not a problem per se, since real populations sizes do fluctuate over time. However, it is often useful to treat the population as a known parameter. This is the case, for example, if we want to predict genomic diversity in a population of known size.
If parents vary in their number of offspring, how can they synchronize to produce exactly one offspring, on average?
One way would be to add feedback, reducing the mean number of offspring when the population size increases, and increasing it when the population size decreases. That could work, but there are many parameters to fix, and even there we can't control the population size all that well.
There are a few elegant but slightly strange ways of solving this problem by keeping the population size at an exact, predefined $N$. First, we'll imagine that reproduction is synchronized across all individuals in the population.
Then, we'll suppose that each parent produces very many offspring, but that only $N$ offspring from the entire population are allowed to survive, and these are selected at random among all offspring. You can also turn the table and imagine that each of the allowed $N$ offspring "picks" a parent at random. If you don't like the idea of an offspring choosing its parents, you can imagine a higher power selecting a parent at random to generate every offspring.
These approaches are equivalent, in that they make the same predictions about the number of surviving offsprings per parent. **Take the time to convince yourself that this is the case.**
This sampling approach defines the Wright-Fisher model. We can add many features to this model, such as selection, recombination, mutation, and so forth, but as long as you have this discrete generations and random selection of parents, you're within the Wright-Fisher model.
I'll just mention one alternative, called the Moran Model, which involves replacing a single, randomly selected individual by the genotype of another, randomly selected individual, and repeating this $N$ times per generation. The Moran model is not exactly equivalent to the Wright-Fisher model, and there are cases where it is more convenient. But we'll stick to Wright-Fisher for this notebook.
*Optional Mathematical exercise*
1. What is the distribution of offspring number per individual in the Wright-Fisher model?
2. Convince yourself that this distribution is approximately Poisson distributed with mean one (hint: This is a consequence of the law of rare events)
## Libraries
We'll need the following python plotting libraries.
<code>
### 1
%matplotlib inline
# If you run into errors with %matplotlib, check that your version of ipython is >=1.0
import numpy as np # Numpy defines useful functions for manipulating arrays and matrices.
import matplotlib.pyplot as plt # Matplotlib is a plotting library
</code>
## Implementation
We have specified almost everything we needed in the model, except for the initial state of the population. We will suppose that the single site has two alleles labeled $0$ and $1$. We need to specify the number of individuals nInd in the initial population and the proportion $p_0$ of $1$ alleles
<code>
### 2
p0 = 0.1 # initial proportion of "1" alleles
nInd = 100 # initial population size (number of individuals)
</code>
Now we need to create an initial population with the given number of individuals, and the appropriate proportion of 1 alleles. We'll store the population as a np.array called "initial_population", of length nInd containing 0s and 1s.
<code>
### 3
# Initialize a population of length nInd with only 0 alleles.
initial_population = np.zeros(nInd)
# Set the first p0*nInd alleles to 1.
......
# Now all the 1 alleles are at the beginning of the array, with the remainder filled with zero.
# The position of individuals doesn't matter in this model, but if you prefer to have a more realistically random
# distribution of alleles, you can use np.random.shuffle to distribute alleles randomly.
</code>
To take finite samples from this population, we can use the np.random.choice function. When taking a sample from a population, we can pick each sample only once--the "replace=False" option below tells us that we don't replace the sampled individual in the population before drawing a new one. Read the np.random.choice documentation for more detail!
<code>
### 4
sample_size = 10
np.random.choice(initial_population, sample_size, replace=False )
</code>
When we take repeated samples from the same population, we can find different numbers of alternate alleles: There are random fluctuations not only in the reproduction, but also in the experimental sampling. We'll first have a look at all these distributions.
*Optional Mathematical exercise*
1-What is the distribution of the number of alternate alleles in a sample of $s$ individuals from a population of size $nInd$ with allele frequency p?
I generated a bunch of samples below, and compared the resulting histogram to plausible probability distributions so that you can pick the one that fits best. If you don't do the math problems, read the wikipedia entry about the best-fitting one and check that it makes sense.
<code>
### 5
import scipy
from scipy import stats
iterations = 10000 # the number of times to draw.
sample_size = 10 # the size of each sample
alt_counts = [] # number of alternate alleles (i.e., 1's) for each draw
for i in range(iterations):
sample = np.random.choice(initial_population, sample_size, replace=False)
# Get the number of alt alleles
alt_counts.append(sample.sum())
# Plot a histogram of sampled values
plt.hist(alt_counts, sample_size + 1, range=(-0.5, sample_size + 1 - 0.5), label="random sample")
plt.xlabel("number of alt alleles")
plt.ylabel("counts")
# Compare this to some discrete distributions
x_range = range(sample_size + 1) # All the possible values
p = np.sum(initial_population) * 1. / len(initial_population) # Initial fraction of alt's
# Poisson with mean sample_size * p
y_poisson = stats.poisson.pmf(x_range, sample_size*p) * iterations
# Binomial with probability p and sample_size draws
y_binom = stats.binom.pmf(x_range, sample_size,p) * iterations
# Hypergeometric draw of sample_size from population of size len(initial_populationpop)
# with np.sum(initial_population) ones.
y_hypergeom = stats.hypergeom.pmf(x_range, len(initial_population), np.sum(initial_population), sample_size)\
* iterations
plt.plot(x_range, y_poisson, label="Poisson", lw=3)
plt.plot(x_range, y_binom, label="Binomial")
plt.plot(x_range, y_hypergeom, label="Hypergeometric")
plt.xlim(-0.5, sample_size + 0.5)
plt.legend()
</code>
Now comes the time to code up the Wright-Fisher model. Remember that there were two ways of thinking about Wright-Fisher reproduction:
1- We generate a very large number of offspring for each parent, and than we take a sample from this very large number of offspring
2- Each offspring picks a parent at random.
In 2-, each parent can be chosen multiple times. This is equivalent to taking a sample from the previous generation, but *with replacement*. Convince yourself that this is true. This is *not* optional! If you are not convinced, try programming it both ways, or ask questions.
Now code a function that takes in a parental population as an array (such as "initial_population", above), and returns an offspring population.
<code>
### 6
def generation(pop):
"""Takes in a list or array describing an asexual parental population.
Return a list describing a descendant population according to Wright-Fisher dynamics with constant size
"""
# Use np.random.choice to draw an offspring population of len(pop) individuals
return np.random.choice(...)
generation(initial_population)
</code>
Here again, we get a different number of ones every time we run the "generation" function. I also generated a bunch of generation samples to get an idea of how much variation there is, and overlaid some plausible distribution. Which one fits best? Does it make sense to you?
<code>
### 7
nsample = 10000 # the number of samples to draw.
alt_counts = [] # number of alternate alleles (i.e., 1's) for each draw
for i in range(nsample):
offspring = generation(initial_population)
alt_counts.append(offspring.sum())
hist = plt.hist(alt_counts, len(initial_population)+1, range=(0-0.5, len(initial_population)+0.5))
plt.xlabel("number of alt alleles")
plt.ylabel("counts")
# Here I just check that the initial population is still a list of length nInd
assert nInd == len(initial_population),"initial_population doesn't have the same length as nInd"
x_range = range(nInd+1) # All the possible values
p=np.sum(initial_population)*1./nInd # The initial frequency
# Compare this to some distributions
y_poisson = stats.poisson.pmf(x_range, nInd*p) * nsample
y_binom = stats.binom.pmf(x_range, nInd, p) * nsample
y_hypergeom = stats.hypergeom.pmf(x_range, nInd, np.sum(initial_population), nInd) * nsample
plt.plot(x_range, y_poisson, label="Poisson",lw=3)
plt.plot(x_range, y_binom, label="Binomial")
plt.plot(x_range, y_hypergeom, label="Hypergeometric")
plt.xlim(-0.5, nInd+0.5)
plt.ylim(0, 1.2*max(hist[0]))
plt.legend()
</code>
Now we are ready to evolve our population for 100 generations. Let's store the entire genotypes for each generation in a list.
<code>
### 8
nGen = 100 # Number of generations to simulate
history = [initial_population] # A container list for our simulations. It will contain the population
# state after generations 0 to nGen
for i in range(nGen):
# evolve the population for one generation, and append the result to history.
history.append(...)
history = np.array(history) # convert the list into an array for convenient manipulation later on
</code>
Now we want to look at the results. Let's compute the allele frequency at each generation and plot that as a function of time.
<code>
### 9
# Compute the allele frequency at each generation.
# freqs should be a list or array of frequencies, with one frequency per generation.
# history is a np array and has two methods that can help you here: sum, and mean.
# Mean is probably the best bet here.
freqs =...
plt.plot(freqs)
plt.axis([0, 100, 0, 1]);#define the plotting range
plt.xlabel("generation")
plt.ylabel("population frequency")
</code>
Now we would like to experiment a bit with the tools that we have developed. Before we do this, we will organize them a bit better, using a Python "class" and object-oriented programming. We have defined above variables that describe a population (such as the population size nInd, and the ancestral frequency g0). We have also defined functions that apply to a population, such as "generation". A class is used to keep track of the relation between objects, variables, and functions.
If you are not familiar with classes and are having issues, have a look at [this tutorial](http://en.wikibooks.org/wiki/A_Beginner's_Python_Tutorial/Classes#Creating_a_Class).
<code>
### 10
class population:
"""
Initialization call:
population(nInd,p0)
requires a number of individuals nInd and an initial frequency p0
Variables:
nInd: The number of individuals
p0: the initial allele frequency
initial_population: an array of nInd alleles
history: a list of genotypes for each generation
frequency_trajectory: an allele frequency trajectory; only defined if get_frequency_trajectory is run.
Methods:
generation: returns the offspring from the current population, whish is also the last one in self.history
evolve: evolves the population for a fixed number of generations, stores results to self.history
get_frequency_trajectory: calculates the allele frequency history for the population
plot_trajectory: plots the allele frequency history for the population
"""
def __init__(self, nInd, p0):
"""initialize the population. nInd is the number of individuals. p0 is the initial allele frequency.
__init__ is a method that, when run, creates a "population" class and defines some of its variables.
Here we define this __init__ method but we don't run it, so there is no "population" created yet.
In the meantime, we'll refer to the eventual population object as "self".
We'll eventually create a population by stating something like
pop = population(nInd,p0)
This will call the __init__ function and pass a "population" object to it in lieu of self.
"""
self.nInd = nInd
self.p0 = p0
# Initialize the population
self.initial_population = np.zeros(self.nInd)
self.initial_population[0 : int(p0*self.nInd)] = 1
np.random.shuffle(self.initial_population)
# History is a container that records the genotype at each generation.
# We'll update this list as we go along. For now it should just contains the initial population
self.history = ...
def generation(self):
"""class methods need "self" as an argument in they definition to know that they apply to a "population"
object. If we have a population "pop", we can write
pop.generation(), and python will know how to pass the population as the first argument.
Returns a descendant population according to Wright-Fisher dynamics with constant size, assuming that the
parental population is the last generation of self.history.
"""
return np....
def evolve(self,nGen):
"""
This is a method with one additional argument, the number of generations nGen.
To call this method on a population "pop", we'd call pop.evolve(nGen).
This function can be called many times on the same population.
pop.evolve(2)
pop.evolve(3)
would evolve the population for 5 generations.
For each step, we make a call to the function generation() and append the population to the "self.history"
container.
"""
for _i in range(nGen):
...
self.get_frequency_trajectory() # This computes self.frequency_trajectory, the list of population frequencies from the first generation
# to the current one. the get_frequency_trajectory function is defined below.
def get_frequency_trajectory(self):
"""
returns an array containing the allele frequency history for the population (i.e., the allele frequency at
generations 0,1,2,..., len(self.history))
"""
history_array = np.array(self.history)
self.frequency_trajectory = ...
return self.frequency_trajectory
def plot_trajectory(self,ax="auto"):
"""
plots the allele frequency history for the population
"""
plt.plot(self.frequency_trajectory)
if ax=="auto":
plt.axis([0, len(self.history), 0, 1])
else:
plt.axis(ax)
</code>
# Exploration
## Drift
We can now define multiple populations, and let them evolve from the same initial conditions.
<code>
### 11
nInd = 50
nGen = 30
nRuns = 30
p0 = 0.02
# Create a list of length nRuns containing initial populations
# with initial frequency p0 and nInd individuals.
pops = [... for i in range(nRuns)]
</code>
Evolve each population for nGen generations. Because each population object has it's own internal storage for the history of the population, we don't have to worry about recording anything.
<code>
### 12
for pop in pops:
...;
</code>
Now plot each population trajectory, using the built-in method from the population class.
<code>
### 13
for pop in pops:
...;
plt.xlabel("generation")
plt.ylabel("population frequency of 1 allele")
</code>
Now that we know it works, let's explore this a bit numerically. Try to get at least 1000 runs, it'll make graphs prettier down the road.
<code>
### 14
nInd = 10
nGen = 10
nRuns = ...
p0 =...
pops = [population(nInd, p0) for i in range(nRuns)]
for pop in pops:
...
...
plt.xlabel("generation")
plt.ylabel("population frequency")
</code>
So there is a lot of randomness in there, but if you run it multiple times you should see that there is some regularity in how fast the allele frequencies depart from the initial values.
To investigate this, calculate and plot the distribution of frequency at each generation.
<code>
### 15
def frequencyAtGen(generation_number, populations, nBins=11):
"""calculates the allele frequency at generation genN for a list of populations pops.
Generates a histogram of the observed values"""
counts_per_bin, bin_edge_positions = np.histogram([... for pop in populations], bins=nBins, range=(0,1))
bin_centers=np.array([(bin_edge_positions[i+1]+bin_edge_positions[i]) / 2 for i in range(len(counts_per_bin))])
return bin_centers, counts_per_bin # Return the data from which we will generate the plot
</code>
<code>
### 16
nBins = 11 # The number of frequency bins that we will use to partition the data.
for i in range(nGen+1):
bin_centers, counts_per_bin = frequencyAtGen(...);
if i==0:
plt.plot(bin_centers, counts_per_bin, color=plt.cm.autumn(i*1./nGen), label="first generation")
# cm.autumn(i*1./nGen) returns the
# color with which to plot the current line
elif i==nGen:
plt.plot(bin_centers, counts_per_bin, color=plt.cm.autumn(i*1./nGen), label="generation %d"% (nGen,))
else:
plt.plot(bin_centers, counts_per_bin, color=plt.cm.autumn(i*1./nGen))
plt.legend()
plt.xlabel("Population frequency")
plt.ylabel("Number of simulated populations ")
</code>
There are three important observations here:
1-Frequencies tend to spread out over time
2-Over time, there are more and more populations at frequencies 0 and 1. (Why?)
3-Apart from the 0 and 1 bins, the distribution becomes quite flat.
A few alternate ways of visualizing the data: first a density map
<code>
### 17
nBins = 11
sfs_by_generation = np.array([frequencyAtGen(i, pops, nBins=nBins)[1] for i in range(0, nGen+1)])
bins = frequencyAtGen(i, pops, nBins=nBins)[0]
plt.imshow(sfs_by_generation, aspect=nBins*1./nGen, interpolation='nearest')
plt.xlabel("Population frequency (bin number)")
plt.ylabel("Generation")
plt.colorbar()
</code>
Then a 3D histogram, unfortunately a bit slow to compute.
<code>
### 18
from mpl_toolkits.mplot3d import Axes3D
fig = plt.figure(figsize=(10,10))
ax = fig.add_subplot(111, projection='3d', elev=90)
xedges = bins
yedges = np.arange(nGen+1)
xpos, ypos = np.meshgrid(xedges-.4/nBins, yedges-0.5)
xpos = xpos.flatten()
ypos = ypos.flatten()
zpos = 0 * ypos
dx = .8 / nBins
dy = 1
dz = sfs_by_generation.flatten()
ax.bar3d(xpos, ypos, zpos, dx, dy, dz, color='b', edgecolor='none', alpha=0.15)
ax.view_init(elev=30., azim=60)
ax.set_xlabel("Population frequency")
ax.set_ylabel("Generation")
ax.set_zlabel("Counts")
plt.show()
</code>
Now let's dig into the effect of population size in a bit more detail.
Consider the change in the frequency of alleles between parent and offsprin.
*Mathematical exercise (NOT optional)*:
* What is the distribution of the number of individuals carrying the derived allele after one generation, if the derived allele is at frequency $p$ in a parental population of size $N$?
(Hint: we explored this numerically above!)
* What is the variance of this distribution? (Look it up if you don't know--wikipedia is useful for that kind of stuff).
* What is the variance in the distribution in the derived allele *frequency* (rather than the allele counts)?
To study the effect of population size on the rate of change in allele frequencies, plot the distribution of allele frequencies after nGen generation. Start with nGen=1 generation.
<code>
### 19
histograms = []
variances = []
p0 = 0.2
sizes = [5, 10, 20, 50, 100, 500]
nGen = 1
for nInd in sizes:
pops = [population(nInd, p0) for i in range(1000)]
[pop.evolve(nGen) for pop in pops]
sample = [pop.get_frequency_trajectory()[-1] for pop in pops]
variances.append(np.var(sample))
histograms.append(plt.hist(sample, alpha=0.5, label="size %d" % (nInd,) ))
plt.xlabel("Population frequency")
plt.ylabel("Number of populations")
plt.legend()
</code>
So how does population size affect the change in allele frequency after one generation? Can you give a specific function describing the relationship between variance and population size?
You can get this relationship from the math exercise above, or just try to guess it from the data. If you want to try to guess, start by plotting the variances (stored in "variances") against the population sizes (stored in "sizes"). Then you can either try to plot different functinoal forms to see if they fit, or you can change the way you plot the data such that it looks like a straight line. If you do the latter, make sure you update the labels!
Here I'm giving you a bit more room to explore--there are multiple ways to get there.
<code>
### 20
plt.plot(np.array(sizes), variances, 'o', label="simulation") #this is a starting point, but you can change this!
# Your theory.
......
plt.xlabel("Population size")
plt.ylabel("Variance")
</code>
For short times, the expected changes in allele frequencies, $Var\left[E[(x-x_0)^2)\right]$, are larger for smaller population, a crucial result of population genetics.
The next question is: How does the rate of change in allele frequency depend on the initial allele frequency? We can plot the histograms of allele frequency as before:
<code>
### 21
histograms = []
variances = []
p0_list = np.array([0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, .6, .7, .8, 0.9, 0.95, 1])
nGen = 1
for p0 in p0_list:
pops = [population(nInd, p0) for i in range(1000)]
[pop.evolve(nGen) for pop in pops]
sample = [pop.get_frequency_trajectory()[-1] for pop in pops]
variances.append(np.var(sample))
histograms.append(plt.hist(sample, 100, alpha=0.5, range=(0,1)))
plt.xlabel("Population frequency")
plt.ylabel("Number of populations")
</code>
Find the relationship between initial frequency and variance. You can deduce it from the math exercise above, look it up on wikipedia, but you can also just try to guess it from the simulations.
Tips for guessing:
First, make the plot of variance vs frequency below
Then consider how much variance there is for p0=0 and p0=1.
Can you come up with a simple function that has these values? Hint: it's a very simple function!
<code>
### 22
plt.plot(np.array(p0_list), variances, 'o', label="simulations")
plt.plot(np.array(p0_list), ..., '-', label="theory") # Your theory.
plt.ylabel("Variance")
plt.xlabel(r"initial frequency p_0")
plt.legend()
</code>
Can you explain why this function is symmetrical around $p_0=0.5$?
## Mutation
New mutations enter the population in a single individual, and therefore begin their journey at frequency $\frac{1}{N}$. Next, we'll numerically estimate the probability that such a new mutation will eventually fix (i.e., the probability that the mutation reaches frequency 1) in the population, if no subsequent mutations occur.
<code>
### 23
nInd = 10
nGen = 100
nRuns = 2000
# Enter the initial allele frequency for new mutations
p0 = ...
pops = [... for i in range(nRuns)]
[pop.evolve(nGen) for pop in pops];
</code>
We can plot the number of populations at each frequency, as we did above.
<code>
### 24
nBins = nInd + 1 # We want to have bins for 0,1,2,..., N copies of the allele.
proportion_fixed = [] # fixation rate
for i in range(nGen+1):
x,y = frequencyAtGen(...);
if i==0:
plt.plot(x, y, color=plt.cm.autumn(i*1./nGen), label="first generation") # cm.autumn(i*1./nGen) returns the
#color with which to plot the current line
elif i==nGen:
plt.plot(x, y, color=plt.cm.autumn(i*1./nGen), label="generation %d"% (nGen,) )
else:
plt.plot(x, y, color=plt.cm.autumn(i*1./nGen))
# We'll consider a population "fixed" if it is in the highest-frequency bin. It's
# an approximation, but not a bad one if the number of bins is comparable to the
# population size.
proportion_fixed.append((i, y[-1]*1./nRuns))
plt.legend()
plt.xlabel("Population frequency")
plt.ylabel("Number of simulations")
</code>
Here you should find that most mutations fix at zero frequency--only a small proportion survives.
*What is the probability that a new mutation fixes in the population?*--I'd like you to solve this problem both mathematically and numerically.
The mathematical part requires almost no calculation or mathematical knowledge, once you think about it in the right way. You can include your mathematical solution in the box below.
For the computational part, note that we already computed the proportion of fixed alleles vs time in the "proportion_fixed" variable. So if we simulate long enough, we'll find the proportion of mutations that eventually fix.
Make sure that the numerical value agrees with the mathematical expectation!
<code>
### 25
proportion_fixed = np.array(proportion_fixed)
plt.plot(proportion_fixed[:,0], proportion_fixed[:,1])
plt.xlabel("Generation")
plt.ylabel("Fixation probability")
</code>
# Summary
Some important things that we've seen in this notebook:
* The Wright-Fisher model. Despite its simplicity, it is the basic building block of a large fraction of population genetics.
* In finite populations, sampling fluctuations are an important driver of allele frequency change.
* These sampling fluctuations cause larger frequency changes in smaller populations.
* These fluctuations mean that alleles eventually fix one way or another -- We need new mutations to maintain diversity within a population.
* For neutral alleles, the probability of new mutations fixing in the population is inversely proportional to the population size
# Something to think about.
We'll get to selection, recombination, and linkage in the next exercises. In the meantime, you can think about the following:
* How much time will it take for a typical new mutation to reach fixation for different population sizes?
* If you add a constant influx of new mutations, how will the distribution of allele frequency look like at any given point in time?
Copyright: Simon Gravel. Do not share of distribute this file without the written consent of the author.
|
{
"filename": "wright_fisher-hints_1.ipynb",
"repository": "sparkingdark/dataset-test",
"query": "transformed_from_existing",
"size": 44935,
"sha": ""
}
|
# num-methods-E.ipynb
Repository: subblue/applying-maths-book
# 8 Reaction schemes with feedback. Predator -Prey (Lotka-Volterra) & Nerve impulses (Fitzhugh-Nagumo) equations.
## Introduction
Feedback in a chemical reaction implies that there are at least two reactions for which the product of one is the reactant for the other, and vice versa; for example,
$$\displaystyle X + Y \longrightarrow 2X ,\qquad X \longrightarrow Y$$
Interesting dynamics can be observed in reactions with feedback, because a product that is also a reactant catalyses its own production, i.e. is autocatalytic. There are many such reactions that are now known for example the Belousov - Zhabotinsky ( bromomalonic acid / $\mathrm{Ru^{2+}\,/\,BrO_3^-}$ and traces of $\mathrm{BrO_2}$ ) and chlorine dioxide / I$_2$ / malonic acid reactions, which involve a complex series of coupled chemical reactions inhibiting and also feeding back on one another, and so catalysing the reaction. Many biological reactions rely on feedback, The production of ATP, for example, is produced through a succession of reactions in the glycolytic cycle that involve ATP, thus to produce ATP we need ATP, similarly to produce a cell, a cell is needed.
Feedback is also common in biology, in the interactions of animals with one another, such as the synchronizing of the flashing of fireflies and of the behaviour of predators and prey. In physiology, the electrical response of nerve and cardiac muscle cells show feedback and the physio-chemical system is termed excitable, meaning that under certain specified conditions, far from equilibrium, oscillations in the concentration of different species, or of electrical impulses can occur. See Scott (1995) for a detailed description of oscillating chemical reactions, and Strogatz (1994) for non-linear processes in general. Examples of a few of these processes are now presented.
## 8.1 Predator - prey or Lotka-Volterra equations
One of the simplest set of reactions was first studied by Lotka (1925), it involves two species X and Y where some amount of each is present initially.
$$\displaystyle \qquad\qquad \begin{array}{lll}\\
Y & \overset{k_1}\longrightarrow &Y + Y \\
Y + X& \overset{k_2}\longrightarrow &X + X \\
X & \overset{k_3}\longrightarrow D \\
\end{array}\qquad\qquad \qquad\qquad \text{(48)}$$
The amount of species $Y$ is doubled in the first step and is then lost by reaction with $X$ to produce more $X$ in the second. Initially, $Y$ increases rapidly, but as it does so, the rate of reaction with $X$ increases. This makes more $X$ which accelerates the reaction, and $Y$ is eventually consumed more rapidly than it is formed and its population falls. The population of $X$ then falls, and the process repeats itself.
Later, Volterra (1926), who was studying the variation of animal populations, described the same set of reactions in this way.
"The first case I have considered is that of two associated species, of which one, finding sufficient food in its environment, would multiply indefinitely when left to itself, while the other would perish for lack of nourishment if left alone; but the second feeds upon the first, and so the two species can coexist together. The proportional rate of increase of the prey diminishes as the number of individuals of the predator increases, while the augmentation of the predator increases with the increase of the number of the prey."
The scheme was originally devised to explain the temporal oscillation of the numbers of fish in the Adriatic Sea, however, the longest time series of this oscillatory behaviour appears to be the record of the number of lynx and hare pelts sold by trappers to the Hudson Bay Company in Canada over the period 1848 to 1907. The actual data, while oscillatory, is also rather chaotic, illustrating that the actual behaviour of animals is always going to be rather difficult to model due to the uncontrolled nature of the experiment. Nevertheless, some insight into predator - prey behaviour can be obtained with rate equations which have the general form
$$\displaystyle \text{rate of change}_i = gain_i -loss_i$$
for each species $i = X,Y $. Thus the prey, we associate with species $Y$ grow in the absence of predators as
$$\displaystyle gain_Y=k_1Y$$
and are eaten by predators with a rate proportional by their own number and that of the predators, which is
$$\displaystyle loss_Y=k_2YX$$
Turning our attention to the predators, as they live by eating prey they have a gain proportional to the number of prey and their own number which is $k_2YX$ and their death is only by natural causes thus their loss term is $ k_3X$. Combining these terms gives give the number of each at time $t$ and this enables us to calculate the numbers at the next time $t+1$ as
$$\displaystyle \begin{align}Y_{t+1} &= Y_t + (k_1Y_t-k_2Y_tX_t)\delta t\\
X_{t+1}&=X_t+(k_2Y_tX_t-k_3X_t)\delta t \end{align}$$
where $\delta t $ is the time step. These equations can be numerically integrated but usually we assume that the numbers of animals is large and the time step small compared to the inverse rate constants so that the change $\displaystyle \frac{Y_{t+1}-Y_t}{\delta t}\to \frac{dY}{dt}$ is valid, and similarly for $X$, producing
$$\displaystyle \qquad\qquad\begin{array}{lll}\\
\displaystyle \frac{dY}{dt} & = & k_1Y -k_2 YX & \text{prey}\\
\displaystyle \frac{dX}{dt} & = & k_2 YX - k_3 X& \text{predator}\\
\end{array}\qquad\qquad \qquad\qquad \text{(49)}$$
where $Y$ represents a population of prey and $X$ that of predator. Besides hares and lynx, the creatures could be two types of fish or aphids predated on by ladybirds; you can imagine many other examples.
This model is a great simplification of actual predator - prey interactions; mathematically, the animals are assumed to be so numerous that they can be treated as if they were molecules in a chemical reaction, but more fundamentally, due to the simplicity of their interactions one with another. A more detailed, and necessarily more complex, description is to be found in Britton (2003) and in Murray (2002). Nonetheless, treating the problem as if it were a chemical one, the prey are breeding at a rate $k_1Y$, the mother producing one offspring in each unit of time. The fuel to do this and driving the whole predator - prey scheme is grass, or similar vegetation, assumed to be in unlimited supply. The rate constant $k_1$ is therefore really a pseudo- first-order rate constant and effectively contains a term allowing for the quantity of vegetation available to be eaten. In the second equation the prey is killed at rate $k_2YX$, which allows the predator to breed. The predators die through natural causes at rate $k_3X$; however, all the prey are killed by predators and their population does not die off naturally: there is no term $Y \overset{k_4} \longrightarrow D$, where $k_4$ is the rate of natural deaths. It is assumed in our model for simplicity only that the rate constant for encounter of prey and predator, $k_2$, is the same as the rate of birth of predators, which it may not be. All the rate constants are positive numbers.
In a molecular example, the equations have to be changed slightly; the first step becomes
$$\displaystyle C + Y \overset{k'_1}\longrightarrow Y+Y$$
where $C$ is some compound whose concentration is unchanged during reaction and provides the material and fuel or free energy to form another Y; it is therefore always at vast excess over Y. To make these equations the same as in (11.48), the substitution $k_1 = k'_{1}C$ is made and so $k_1$ is a pseudo, first-order rate constant.
The first step in analysing these equations is to calculate the nullclines and steady state conditions that are also called the equilibrium points. Then the phase plane will be calculated, which plots the number density between predator and prey and finally the time profiles of species $Y$ and $X$. The initial values to do this will be
$$\displaystyle X_0 = 60,\; Y_0 = 100.0,\; k_1 = 1.0,\; k_2 = 0.01,\; k_3 = 0.5,\; t=0\to 40$$
The unit of time could be in seconds or years; this depends on the situation. We need not specify it here, but clearly, the time would be something of the order of a year for hares and lynx. The numerical calculations used to produce the data in Fig. 19, was based on the Euler algorithm (14) with rate equations defined as
<code>
#dydt = lambda y,x : k1*y - k2*y*x # y = prey , x predator eqns 49
#dxdt = lambda y,x : k2*y*x - k3*x}
# in the for loop the increments are
#x = x + h*dxdt(y,x)
#y = y + h*dydt(y,x)
</code>
and 500 points were used in the normal Euler method of integration.

Figure 19. The time profiles of the predator $X$ and prey $Y$. The prey's population rises first; the predator population always lags behind. The small circle is at $t = 2$ and is shown in Figure 20. $X_0 = 60,\; Y_0 = 100.0,\; k_1 = 1.0,\; k_2 = 0.01,\; k_3 = 0.5$.
____
The time profiles, Fig. 19, show that the prey population rises before that of the predator. Initially, the prey (hares) breeds and its population increases exponentially, and would continue to do so if it were not for predators because we assume in our limited model that hares do not die naturally and there is always plenty of grass for them to eat. As the predator (lynx) kills its prey (hares) so that it may thrive, the prey's population growth is limited by the number of predators and their success in catching them, so that their population reaches a maximum, and starts to fall. This is due to the hares breeding and so increasing in number but these being decreased by predation. However, the predator population has now grown too large, and as there is now less prey to support such numbers, the predator population falls because these die off by natural causes determined by rate constant $k_3$. Since the predator population has fallen, the prey population, fed on an everlasting amount of grass, can now recover and their population increases and so the sequence repeats itself.
### **Steady State**
The steady state conditions are found when the rate of change of each population is zero;
$$\displaystyle \frac{dY}{dt} = k_1Y -k_2 YX=0 \qquad \frac{dX}{dt} = k_2 YX - k_3 X=0 $$
giving $Y = X = 0$ as one solution and $Y_e = k_3/k_2,\; X_e = k_1/k_2$ as the other. The nullclines are the equations produced when the derivatives are zero, and in this instance, they are the horizontal and vertical straight lines crossing the axes at the equilibrium values, $Y_e$ and $X_e$. In more complex sets of equations the nullclines could be curves. In Figure 20, the nullclines are plotted as dashed lines. The first equilibrium point at the origin is obvious; no prey and no predator. The second at, $X_e = 50,\; Y_e = 100$ means that the populations can remain stable given these ratios of rate constants. If the populations are initially different, then they will oscillate in value, *ad infinitum*, to a greater or lesser extent.
To obtain the phase plane equation, the rate of change of the predator with prey is needed. This is
$$\displaystyle \frac{dX}{dY}=\frac{\displaystyle\frac{dX}{dt}}{\displaystyle\frac{dY}{dt}} = \frac{(k_2Y-k_3)X}{(k_1-k_2X)Y}$$
Separating variables before integrating gives
$$\displaystyle \int\frac{k_2Y-k_3}{Y}dY=\int\frac{k_1-k_2X}{X}dX$$
which evaluates to
$$\displaystyle k_2Y-k_3\ln(Y)=k_1\ln(X)-k_2X+C$$
with an arbitrary constant $C$. Although this equation does not explicitly contain time, $X$ and $Y$ do change with time. The constant $C$ is evaluated from the initial conditions and the curve produced moves around the non-zero stable point in an anticlockwise manner.
You can see that the last equation above describing the phase-plane is a rather hard one because it is transcendental. We want to plot $Y$ vs $X$, but the equation cannot easily be put in the form $Y = \cdots$. Numerically solving the equation $k_2Y - k_3\ln(Y) = Q$ for each value of $X$ where $Q$ is the value of the right-hand side is very tedious, and is made difficult because the equation produces real as well as complex solutions. The simplest way to draw the phase plane curve is therefore to plot the populations for a particular set of parameters. If EulerX and EulerY are the arrays containing the $X$ and $Y$ populations calculated with the Euler method algorithm, then the instructions to plot, say, the first 120 points are shown below with the point at time 2 also plotted as a circle. It is assume that the number of points, $n$ and $maxt$ are already defined.
<code>
#plt.plot(Eulery[0:120],Eulerx[0:120])
#tt= int(2*n/maxt)
#plt.scatter(Eulery[tt],Eulerx[tt],s=100)
</code>
The phase-plane curve is closed and orbits the steady state or equilibrium point $(X_e,\, Y_e)$, which means that the oscillations in population continue for ever. If several different pairs of starting population are used, a series of separate closed curves are produced on the phase plane arranged one inside the other, as if they were contours drawn on the inside of a bowl with the equilibrium point at the centre.
The nullclines clearly split the phase plane into four areas and separate populations at their maxima or minima, Fig. 20. In the bottom right quadrant containing the initial population, both predator and prey populations increase. When the prey reaches a maximum, the $X = 100$ nullcline is crossed vertically, and the predator population increases while the prey decreases. This quadrant contains the point that is shown in Fig.19 as the small circle. At the $Y = 50$ vertical nullcline, the predator population peaks. In the next quadrant (top left), both predator and prey population decrease, and in the final quadrant the predator continues to decrease while the prey recovers. It is interesting to note that the average predator lifetime, in the absence of food from prey, is very short at $2$ units of time, which is shown on Fig. 19.

Figure 20. Phase-plane plot with the parameters $X_0 = 60,\; Y_0 = 100,\; k_1 = 1, \;k_2 = 0.01,\; k_3 = 0.5$.
$X$ represents the predator, $Y$ the prey. The equilibrium and initial values are labelled. The circle at $t = 2$ and that at time zero shows that time moves anticlockwise on this plot. (Note that $X$ is plotted on the vertical axis.)
___________
The closed form of the phase plane curve shows that the populations oscillate periodically, and will continue to do so for ever. If different initial conditions apply, then a similar plot is produced but with larger or smaller amplitudes. In fact there are an infinite number of these all circling the stable point, $X_e,\; Y_e$. This behaviour is in contrast to the limit-cycle behaviour of some oscillating reactions, as observed with the Fitzhugh - Nagumo equations, Section 8.2.
This simple model only gives an indication of what may happen between predator and prey. It is a starting point from which a number of interesting questions can be asked about how animals interact in a more realistic way or even as to how ecosystems behave. One simple change to the model is to limit the amount of grass available to the prey, and hence to their total population in the absence of predators.
## 8.3 Nerve impulses and the Fitzhugh - Nagumo equations
The biological cell membrane has a potential difference between its inner and outer surfaces. This potential, along with a pH difference, is used by the molecular motor protein ATPase, either to phosphorylate ADP to ATP, or to hydrolyse ATP to ADP. The membrane in its simplest form can be described as a capacitor and resistor in parallel. However, the membrane's electrical properties are not passive but excitable, which means that if a current impulse above a certain limit is applied, the membrane potential subsequently oscillates continuously. From 1948 to 1952, Hodgkin and Huxley conducted experiments on the axon of the giant squid. These 'patch-clamp' experiments were analysed by assuming that channels for Na$^+$ and K$^+$ ions existed and that the resistance of the axon was voltage dependent. They modelled this behaviour with four coupled differential equations. Their work, together with that of Eccles received the 1963 Nobel Prize for Medicine.
This model of the membrane was itself subsequently parameterised by Fitzhugh who simplified it into two differential equations. This was possible because different ions transport on different time scales, Na$^+$ being slow, and rates could be separated on this basis. The equations in reduced form become
$$\displaystyle \begin{array}{lll}\\
\displaystyle \frac{dv}{dt} & = & v(1-v)(v-\alpha)-w+C \\
\displaystyle \frac{dw}{dt} & = & \epsilon(v-\gamma w)\\
\end{array}$$
The potential $v$ is the fast responding voltage; $w$ is the slow (Na$^+$) recovery one; $\alpha,\; \epsilon, \; \gamma$ are constants with $0 < \alpha < 1,\; \epsilon \ll 1$; and $C$ is an optional applied current. The $v$ nullcline is the cubic equation
$$\displaystyle w = v(1 - v)(v - \alpha) + C$$
the $w$ nullcline is a straight line
$$\displaystyle w = v/\gamma $$
These are plotted in Figure 21 as dotted lines with the parameters, $\alpha = 0.1,\; \epsilon = 0.02$, and $\gamma = 2$. The starting values for the calculation of the phase plane were $w_0 = -0.2,\; v_0 = 0.25$, and $C = 0$, and in Figure 22, $C = 0.025$.
The equilibrium point where the nullclines meet is the solution of
$$\displaystyle v - \gamma v(1 - v)(v - \alpha) - \gamma C = 0$$
which is a cubic and has an algebraic solution of great complexity but which is easily evaluated when the constants are given values. In Figure 21, the applied current $C$ is zero and the response of the nerve is to produce one spike and then a highly damped oscillation in $v$ and $w$ signals. The $v$ and $w$ signals are calculated numerically using the Euler method, Algorithm 14 and more accurately, with Algorithm 15.
The phase plane shows that the $v$ signal initially increases far more rapidly than does $w$, then $v$ decreases slowly as $w$ increases following the nullcline, but the response breaks close to the maximum in the $v$ nullcline. The response then jumps to the other branch of the nullcline that is then followed back to zero about which a few oscillations occur before reaching the stable point. The oscillations are small and difficult to see on the scale of the plot.
When $C$ is not zero, the $v$ nullcline is raised and now instead of oscillating about the stable point where the nullclines meet, a limit-cycle is produced and the phase plane continuously cycles the stable point. A limit-cycle means that the parameters controlling the rate equations are such that the same closed curve is produced whatever the initial $v$ and $w$ values are. This is an important result because the oscillation frequency becomes a well defined function of the physio-chemical state of the system, whereas in the Lotka - Volterra case the oscillation frequency is arbitrary and changes with the initial conditions given to the differential equations.
When the $v$ nullcline is raised up sufficiently by a large value of $C$, the $w$ nullcline crosses it past its maximum and now the new stable point is formed here rather than near to the lowest point and no oscillations occur; this is not shown and is left as a problem. The limit-cycle is formed when the (stable) point formed, which is where the nullclines cross, is between the maximum and minimum of the $v$ nullcline.
The current $C$ was constant in these examples, but it can be pulsed or made into two pulses separated in time, in which case, chaotic behaviour can be produced. A full discussion can be found in Murray (2002).

Figure 21 FitzHugh–Nagumo equation's phase plane plot and time profile with $C = 0$ and other values as in the text. The trajectory focuses onto the equilibrium point (0,0) and oscillated about this point although it appears to end here.
____

Figure 22 FitzHugh–Nagumo equations phase plane plot and time profile with $C = 0.05$ and other values as in the text. The excitable medium in this case produces a limit-cycle. An equilibrium point occurs where the nullclines cross and is not reached by the trajectory following the limit-cycle.
________
## 8.4 Limit-cycles
In Figures 21 and 22, depending on the starting conditions, the trajectory focuses on the equilibrium point or studiously avoids it. The latter produces a limit-cycle that can most simply be understood by considering the mechanical analogy of a harmonic vs a double well potential. In one dimension they can both be represented by
$$\displaystyle V(x) = ax^2 + bx^4$$
The harmonic potential has $a \ge 0$; the double well $a \lt 0$, Figure 23. Now suppose that these represent a profile cut through a cylindrical surface formed by rotating about the vertical. The trajectory of a ball released anywhere and at any angle on the harmonic surface, $a \ge 0$, will always reach the minimum. In the double well potential, which has the same equation, but with $a \lt 0$, has a minimum that forms a valley running around the bottom of the potential and any trajectory will end up here. The path around the minimum is the equivalent to the path followed in the limit-cycle; no matter where the trajectory starts from it will spiral around and end up following the same path. By changing one parameter, a stable equilibrium becomes an unstable one, the former equilibrium position is avoided, and completely different behaviour is observed. The idea of *bifurcation* now arises naturally, because, by changing one parameter a stable point splits into two, or bifurcates, forming an unstable point and two stable ones. This was met in a mathematical sense with the logistic equation, see Section 1.4.

Figure 23. Two potentials with the form $V(x) = ax^2 + bx^4$.The parabola has $b=0$ and the double well with $b \gt 0$ but with $a$ negative. The double well has both unstable and stable points. The stable points are indicated by the filled circles.
|
{
"filename": "num-methods-E.ipynb",
"repository": "subblue/applying-maths-book",
"query": "transformed_from_existing",
"size": 25861,
"sha": ""
}
|
# R-api.ipynb
Repository: kipoi/kipoi
# Using Kipoi from R
Thanks to the [reticulate](https://github.com/rstudio/reticulate) R package from RStudio, it is possible to easily call python functions from R. Hence one can use kipoi python API from R. This tutorial will show how to do that.
Make sure you have git-lfs and Kipoi correctly installed:
1. Install git-lfs
- `conda install -c conda-forge git-lfs && git lfs install` (alternatively see <https://git-lfs.github.com/>)
2. Install kipoi
- `pip install kipoi`
Please read [docs/using/getting started](http://kipoi.org/docs/using/01_Getting_started/) before going through this notebook.
## Install and load `reticulate`
Make sure you have the reticulate R package installed
<code>
# install.packages("reticulate")
</code>
<code>
library(reticulate)
</code>
## Reticulate quick intro
In general, using Kipoi from R is almost the same as using it from Python: instead of using `object.method()` or `object.attribute` as in python, use `$`: `object$method()`, `object$attribute`.
<code>
# short reticulate example
os <- import("os")
os$chdir("/tmp")
os$getcwd()
</code>
### Type mapping R <-> python
Reticulate translates objects between R and python in the following way:
| R | Python | Examples |
|------------------------|-------------------|---------------------------------------------|
| Single-element vector | Scalar | `1`, `1L`, `TRUE`, `"foo"` |
| Multi-element vector | List | `c(1.0, 2.0, 3.0)`, `c(1L, 2L, 3L)` |
| List of multiple types | Tuple | `list(1L, TRUE, "foo")` |
| Named list | Dict | `list(a = 1L, b = 2.0)`, `dict(x = x_data)` |
| Matrix/Array | NumPy ndarray | `matrix(c(1,2,3,4), nrow = 2, ncol = 2)` |
| Function | Python function | `function(x) x + 1` |
| NULL, TRUE, FALSE | None, True, False | `NULL`, `TRUE`, `FALSE` |
For more info on reticulate, please visit https://github.com/rstudio/reticulate/.
## Setup the python environment
With `reticulate::py_config()` you can check if the python configuration used by reticulate is correct. You can can also choose to use a different conda environment with `use_condaenv(...)`. This comes handy when using different models depending on different conda environments.
<code>
reticulate::py_config()
</code>
List all conda environments:
```R
reticulate::conda_list()
```
Create a new conda environment for the model:
```
$ kipoi env create HAL
```
Use that environment in R:
```R
reticulate::use_condaenv("kipoi-HAL')
```
## Load kipoi
<code>
kipoi <- import("kipoi")
</code>
### List models
<code>
kipoi$list_models()$head()
</code>
`reticulate` currently doesn't support direct convertion from `pandas.DataFrame` to R's `data.frame`. Let's make a convenience function to create an R dataframe via matrix conversion.
<code>
#' List models as an R data.frame
kipoi_list_models <- function() {
df_models <- kipoi$list_models()
df <- data.frame(df_models$as_matrix())
colnames(df) = df_models$columns$tolist()
return(df)
}
</code>
<code>
df <- kipoi_list_models()
</code>
<code>
head(df, 2)
</code>
### Get the kipoi model and make a prediction for the example files
To run the following example, make sure you have all the dependencies installed. Run:
```R
kipoi$install_model_requirements("MaxEntScan/3prime")
```
from R or
```bash
kipoi env create MaxEntScan
source activate kipoi-MaxEntScan
```
from the command-line. This will install all the required dependencies for both, the model and the dataloader.
<code>
kipoi$install_model_requirements("MaxEntScan/3prime")
</code>
<code>
model <- kipoi$get_model("MaxEntScan/3prime")
</code>
<code>
predictions <- model$pipeline$predict_example()
</code>
<code>
head(predictions)
</code>
### Use the model and dataloader independently
<code>
# Get the dataloader
setwd('~/.kipoi/models/MaxEntScan/3prime')
dl <- model$default_dataloader(gtf_file='example_files/hg19.chr22.gtf', fasta_file='example_files/hg19.chr22.fa')
</code>
<code>
# get a batch iterator
it <- dl$batch_iter(batch_size=4)
</code>
<code>
it
</code>
<code>
# Retrieve a batch of data
batch <- iter_next(it)
</code>
<code>
str(batch)
</code>
<code>
# make the prediction with a model
model$predict_on_batch(batch$inputs)
</code>
## Troubleshooting
Since Kipoi is not natively implemented in R, the error messages are cryptic and hence debugging can be a bit of a pain.
### Run the same code in python or CLI
When you encounter an error, try to run the analogous code snippet from the command line or python. A good starting point is to first run
```
$ kipoi test MaxEntScan/3prime --source=kipoi
```
from the command-line first.
### Dependency issues
It's very likely that the error will be due to missing dependencies. Also note that some models will work only with python 3 or python 2. To install all the required dependencies for the model, run:
```
$ kipoi env create MaxEntScan
$ source activate kipoi-MaxEntScan
```
This will install the dependencies into your current conda environment. If you wish to create a new environment with all the dependencies installed, run
```
$ kipoi env create MaxEntScan
$ source activate kipoi-MaxEntScan
```
To use that environment in R, run:
```R
use_condaenv("kipoi-MaxEntScan__3prime")
```
Make sure you run that code snippet right after importing the `reticulate` library (i.e. make sure you run it before `kipoi <- import('kipoi')`)
### Float/Double type issues
When using a pytorch model: `DeepSEA/predict`
<code>
kipoi$install_model_requirements("DeepSEA/predict")
</code>
<code>
# Get the dataloader
setwd('~/.kipoi/models/DeepSEA/predict')
model <- kipoi$get_model("DeepSEA/predict")
dl <- model$default_dataloader(intervals_file='example_files/intervals.bed', fasta_file='example_files/hg38_chr22.fa')
# get a batch iterator
it <- dl$batch_iter(batch_size=4)
# predict for a batch
batch <- iter_next(it)
</code>
<code>
# model$predict_on_batch(batch$inputs)
</code>
We get an error:
```
Error in py_call_impl(callable, dots$args, dots$keywords): RuntimeError: Input type (CUDADoubleTensor) and weight type (CUDAFloatTensor) should be the same
```
This means that the feeded array is Double instead of Float.
R arrays are by default converted to float64 numpy dtype:
<code>
np <- import("numpy", convert=FALSE)
np$array(0.1)$dtype
</code>
<code>
np$array(batch$inputs)$dtype
</code>
To fix this, we need to explicitly convert them to `float32` before passing the batch to the model:
<code>
model$predict_on_batch(np$array(batch$inputs, dtype='float32'))
</code>
|
{
"filename": "R-api.ipynb",
"repository": "kipoi/kipoi",
"query": "transformed_from_existing",
"size": 62325,
"sha": ""
}
|
# Deseq2_R.ipynb
Repository: Mangul-Lab-USC/RNA-SEQ-Tutorial-PART1
<code>
?system
version
</code>
---
Analyze gene count data using Deseq2
---
<code>
install.packages("rgl", repos = "http://cran.rstudio.com/")
install.packages("ConsRank", repos = "http://cran.rstudio.com/")
library("ConsRank")
</code>
<code>
system("add-apt-repository -y ppa:marutter/rrutter")
system("add-apt-repository -y ppa:marutter/c2d4u")
system("apt-get update")
system("apt install -y r-cran-rstan")
</code>
<code>
install.packages("ggplot2")
</code>
<code>
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
</code>
<code>
BiocManager::install(version="3.9", ask=FALSE)
</code>
<code>
BiocManager::install("DESeq2", version="3.9")
</code>
<code>
# Download the Data
</code>
<code>
countsName <- "https://bioconnector.github.io/workshops/data/airway_scaledcounts.csv"
download.file(countsName, destfile = "airway_scaledcounts.csv", method = "auto")
countData <- read.csv('airway_scaledcounts.csv', header = TRUE, sep = ",")
head(countData)
</code>
<code>
metaDataName <- "https://bioconnector.github.io/workshops/data/airway_metadata.csv"
download.file(metaDataName, destfile = "airway_metadata.csv", method = "auto")
metaData <- read.csv('airway_metadata.csv', header = TRUE, sep = ",")
metaData
# metadata matrix: an matrix in the form of pandas dataframe,
# see DESeq2 manual, samplenames as rownames following treatment type (A,B)
# sampleA1 A
# sampleA2 A
# sampleB1 B
# sampleB2 B
</code>
**Differential analysis Calculation with Salmon data using DESEQ2**
Construct DESEQDataSet Object
<code>
library(ggplot2)
library( "DESeq2" )
</code>
<code>
dds <- DESeqDataSetFromMatrix(countData=countData,
colData=metaData,
design=~dex, tidy = TRUE)
</code>
<code>
#Design specifies how the counts from each gene depend on our variables in the metadata
#For this dataset the factor we care about is our treatment status (dex)
#tidy=TRUE argument, which tells DESeq2 to output the results table with rownames as a first #column called 'row.
#let's see what this object looks like
dds
</code>
Run DESEQ function
<code>
dds <- DESeq(dds)
#estimateSizeFactors
#This calculates the relative library depth of each sample
#estimateDispersions
#estimates the dispersion of counts for each gene
#nbinomWaldTest
#calculates the significance of coefficients in a Negative Binomial GLM using the size and dispersion outputs
</code>
Results Table
<code>
res <- results(dds, tidy=TRUE)
head(res)
</code>
Summary of differential gene expression
<code>
summary(res)
</code>
Sort summary list by p-value
<code>
res <- res[order(res$padj),]
head(res)
</code>
Plot Counts
<code>
#we can use plotCounts fxn to compare the normalized counts
#between treated and control groups for our top 6 genes
par(mfrow=c(2,3))
plotCounts(dds, gene="ENSG00000152583", intgroup="dex")
plotCounts(dds, gene="ENSG00000179094", intgroup="dex")
plotCounts(dds, gene="ENSG00000116584", intgroup="dex")
plotCounts(dds, gene="ENSG00000189221", intgroup="dex")
plotCounts(dds, gene="ENSG00000120129", intgroup="dex")
plotCounts(dds, gene="ENSG00000148175", intgroup="dex")
#Next steps in exploring these data...BLAST to database to find associated gene function
</code>
Volcano Plot
<code>
library(repr)
options(repr.plot.width=10, repr.plot.height=7)
</code>
<code>
#reset par
par(mfrow=c(1,1))
# Make a basic volcano plot
with(res, plot(log2FoldChange, -log10(pvalue), pch=20, main="Volcano plot", xlim=c(-3,3)))
# Add colored points: blue if padj<0.01, red if log2FC>1 and padj<0.05)
with(subset(res, padj<.01 ), points(log2FoldChange, -log10(pvalue), pch=20, col="blue"))
with(subset(res, padj<.01 & abs(log2FoldChange)>2), points(log2FoldChange, -log10(pvalue), pch=20, col="red"))
</code>
<code>
#First we need to transform the raw count data
#vst function will perform variance stabilizing transformation
vsdata <- vst(dds, blind=FALSE)
</code>
<code>
plotPCA(vsdata, intgroup="dex")
#using the DESEQ2 plotPCA fxn we can look at how our samples group by treatment
</code>
|
{
"filename": "Deseq2_R.ipynb",
"repository": "Mangul-Lab-USC/RNA-SEQ-Tutorial-PART1",
"query": "transformed_from_existing",
"size": 214471,
"sha": ""
}
|
# Get_ICD_11.ipynb
Repository: dkisselev-zz/mmc-pipeline
<a href="https://colab.research.google.com/github/dkisselev-zz/mmc-pipeline/blob/main/Get_ICD_11.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a>
<code>
!pip install pymed google-generativeai boto3
</code>
<code>
from pymed import PubMed
import pandas as pd
import google.generativeai as genai
from google.colab import userdata
import requests
import time
import json
import time
from typing import List, Dict, Optional, Tuple
import re
import xml.etree.ElementTree as ET
import boto3
from botocore.client import Config
from botocore import UNSIGNED
from botocore.exceptions import NoCredentialsError, ClientError
</code>
<code>
try:
GEMINI_API_KEY = userdata.get('GOOGLE_API_KEY')
genai.configure(api_key=GEMINI_API_KEY)
except (ValueError, FileNotFoundError):
raise ValueError("GOOGLE_API_KEY not found in Colab secrets. Please add it.")
try:
ICD11_CLIENT_ID = userdata.get('ICD11_CLIENT_ID')
ICD11_CLIENT_SECRET = userdata.get('ICD11_CLIENT_SECRET')
except (ValueError, FileNotFoundError):
raise ValueError("ICD11 not found in Colab secrets. Please add it.")
try:
EMAIL = userdata.get('EMAIL')
except (ValueError, FileNotFoundError):
raise ValueError("EMAIL not found in Colab secrets. Please add it.")
</code>
<code>
class DOIClassifier:
def __init__(self, gemini_api_key: str, icd11_client_id: Optional[str] = None, icd11_client_secret: Optional[str] = None):
"""
Initialize the DOI classifier with Gemini API key and optional ICD-11 OAuth credentials.
Args:
gemini_api_key (str): Google Gemini API key
icd11_client_id (Optional[str]): WHO ICD-11 API client ID
icd11_client_secret (Optional[str]): WHO ICD-11 API client secret
"""
self.pubmed = PubMed(tool="DOIClassifier", email=EMAIL)
genai.configure(api_key=gemini_api_key)
self.model = genai.GenerativeModel('gemini-1.5-flash')
# ICD-11 API endpoint and OAuth credentials
self.icd11_api_base = "https://id.who.int/icd/entity"
self.icd11_token_url = "https://icdaccessmanagement.who.int/connect/token"
self.icd11_client_id = icd11_client_id
self.icd11_client_secret = icd11_client_secret
self.icd11_access_token = None
self.icd11_token_expires_at = None
# Set up headers for ICD-11 API calls
self.icd11_headers = {
'Accept': 'application/json',
'API-Version': 'v2',
'Accept-Language': 'en'
}
# Get initial access token if credentials are provided
if self.icd11_client_id and self.icd11_client_secret:
self._get_icd11_access_token()
def _get_icd11_access_token(self):
"""Request an access token from WHO ICD-11 API using OAuth 2.0."""
try:
token_data = {
'grant_type': 'client_credentials',
'client_id': self.icd11_client_id,
'client_secret': self.icd11_client_secret,
'scope': 'icdapi_access'
}
response = requests.post(self.icd11_token_url, data=token_data)
response.raise_for_status()
token_info = response.json()
self.icd11_access_token = token_info.get('access_token')
expires_in = token_info.get('expires_in', 3600) # Default to 1 hour
# Calculate expiration time (subtract 5 minutes for safety)
self.icd11_token_expires_at = time.time() + expires_in - 300
if self.icd11_access_token:
self.icd11_headers['Authorization'] = f'Bearer {self.icd11_access_token}'
print("Successfully obtained ICD-11 API access token")
else:
print("Warning: Could not obtain ICD-11 access token")
except Exception as e:
print(f"Error obtaining ICD-11 access token: {str(e)}")
self.icd11_access_token = None
def _ensure_valid_icd11_token(self):
"""Ensure we have a valid access token, refresh if needed."""
if not self.icd11_client_id or not self.icd11_client_secret:
return
current_time = time.time()
# Check if token is expired or will expire soon
if (not self.icd11_access_token or
not self.icd11_token_expires_at or
current_time >= self.icd11_token_expires_at):
self._get_icd11_access_token()
def read_dois_from_file(self, file_path: str) -> List[str]:
"""
Read DOIs from a newline-separated file.
Args:
file_path (str): Path to the file containing DOIs
Returns:
List[str]: List of DOIs
"""
with open(file_path, 'r') as file:
dois = [line.strip() for line in file if line.strip()]
return dois
def get_article_info(self, doi: str) -> Optional[Dict]:
"""
Retrieve article title and abstract using pymed.
Args:
doi (str): DOI of the article
Returns:
Optional[Dict]: Dictionary containing title and abstract, or None if not found
"""
try:
# Search for the article using DOI
query = f"{doi}[doi]"
results = self.pubmed.query(query, max_results=1)
for article in results:
meta_xml = getattr(article, 'xml', '')
title = self.get_clean_article_title_from_xml(meta_xml)
abstract = self.get_clean_abstract_from_xml(meta_xml)
if title or abstract:
return {
'doi': doi,
'title': title,
'abstract': abstract
}
return None
except Exception as e:
print(f"Error retrieving article for DOI {doi}: {str(e)}")
return None
def classify_disease_with_gemini(self, title: str, abstract: str) -> Dict:
"""
Use Gemini to classify the disease mentioned in the article.
Args:
title (str): Article title
abstract (str): Article abstract
Returns:
Dict: Dictionary containing disease name and confidence
"""
prompt = f"""
Analyze the following research article and identify the primary disease or medical condition being investigated.
Title: {title}
Abstract: {abstract}
Please provide your response in the following JSON format. The "disease_name" should be the full, unabbreviated name and must not contain any acronyms or parentheses.
{{
"disease_name": "The specific disease or condition name",
"confidence": "high/medium/low",
"reasoning": "Brief explanation of why this disease was identified"
}}
Focus on the most specific disease name mentioned. If multiple diseases are mentioned, choose the primary one being investigated.
"""
# For the 'disease_name', provide only the full name, completely spelled out. For example, if the text mentions "Myocardial Infarction (MI)", the value should be "Myocardial Infarction", not "MI" or "Myocardial Infarction (MI)".
try:
response = self.model.generate_content(prompt)
# Extract JSON from response
response_text = response.text
json_match = re.search(r'\{.*\}', response_text, re.DOTALL)
if json_match:
result = json.loads(json_match.group())
return result
else:
return {
"disease_name": "Unknown",
"confidence": "low",
"reasoning": "Could not parse Gemini response"
}
except Exception as e:
print(f"Error classifying disease: {str(e)}")
return {
"disease_name": "Unknown",
"confidence": "low",
"reasoning": f"Error: {str(e)}"
}
def search_icd11_code(self, disease_name: str) -> Dict:
"""
Search for ICD-11 code and description for a given disease.
Args:
disease_name (str): Name of the disease
Returns:
Dict: Dictionary containing ICD-11 code and description
"""
try:
# Ensure we have a valid access token before making API calls
self._ensure_valid_icd11_token()
# Step 1: Search for foundation entities
search_url = f"https://id.who.int/icd/entity/search"
params = {
'q': disease_name,
# 'propertiesToBeSearched': 'Title,Definition,Exclusion,FullySpecifiedName',
# 'useFlexisearch': 'true',
'flatResults': 'true'
}
response = requests.get(search_url, params=params, headers=self.icd11_headers)
response.raise_for_status()
data = response.json()
if data.get('destinationEntities'):
entity = data['destinationEntities'][0]
entity_url = entity.get('id') # This is the full URL like "http://id.who.int/icd/entity/359051131"
# Extract numeric ID from the entity URL
entity_id_match = re.search(r'/entity/(\d+)$', entity_url)
if not entity_id_match:
return {
'icd11_code': 'Error',
'icd11_description': 'Could not extract entity ID from URL',
'search_confidence': 'error'
}
entity_id = entity_id_match.group(1)
# Step 2: Get linearization data to get the actual ICD-11 code
# Use latest MMS release (2025-01) and extract numeric ID from entity URI
linearization_url = f"https://id.who.int/icd/release/11/2025-01/mms/{entity_id}"
linearization_response = requests.get(linearization_url, headers=self.icd11_headers)
if linearization_response.status_code == 200:
linearization_data = linearization_response.json()
icd11_code = linearization_data.get('code', '')
title_info = linearization_data.get('title', {})
icd11_description = title_info.get('@value', '') if isinstance(title_info, dict) else str(title_info)
return {
'icd11_code': icd11_code,
'icd11_description': icd11_description,
'search_confidence': 'high'
}
return {
'icd11_code': 'Not found',
'icd11_description': 'No ICD-11 code found for this disease',
'search_confidence': 'low'
}
except Exception as e:
print(f"Error searching ICD-11 for {disease_name}: {str(e)}")
return {
'icd11_code': 'Error',
'icd11_description': f'Error searching ICD-11: {str(e)}',
'search_confidence': 'error'
}
def process_dois(self, doi_file_path: str, output_file: str = 'article_classification.csv'):
"""
Process all DOIs in the file and save results to CSV.
Args:
doi_file_path (str): Path to file containing DOIs
output_file (str): Output CSV file path
"""
dois = self.read_dois_from_file(doi_file_path)
results = []
print(f"Processing {len(dois)} DOIs...")
for i, doi in enumerate(dois, 1):
print(f"Processing DOI {i}/{len(dois)}: {doi}")
# Get article information
article_info = self.get_article_info(doi)
if article_info:
# Classify disease
disease_info = self.classify_disease_with_gemini(
article_info['title'],
article_info['abstract']
)
# Search ICD-11 code
icd11_info = self.search_icd11_code(disease_info['disease_name'])
# Combine results
result = {
'doi': doi,
'title': article_info['title'],
'abstract': article_info['abstract'],
'disease_name': disease_info['disease_name'],
'classification_confidence': disease_info['confidence'],
'classification_reasoning': disease_info['reasoning'],
'icd11_code': icd11_info['icd11_code'],
'icd11_description': icd11_info['icd11_description'],
'icd11_search_confidence': icd11_info['search_confidence']
}
results.append(result)
# Add delay to avoid rate limiting
time.sleep(1)
else:
print(f"Could not retrieve information for DOI: {doi}")
# Save to CSV
df = pd.DataFrame(results)
df.to_csv(output_file, index=False)
print(f"Results saved to {output_file}")
return df
@staticmethod
def get_clean_article_title_from_xml(article_xml_element):
"""Extracts and cleans the article title from the XML."""
if not isinstance(article_xml_element, ET.Element): return "N/A"
try:
title_element = article_xml_element.find('.//ArticleTitle')
if title_element is not None:
# Reconstruct text content, handling tags like <i>, <b>
raw_title = "".join(title_element.itertext())
# Clean up whitespace
clean_title = re.sub(r'\s+', ' ', raw_title).strip()
return clean_title
return "N/A"
except Exception as e:
print(f"Error extracting title from XML: {e}")
return "Error"
@staticmethod
def get_clean_abstract_from_xml(article_xml_element):
"""
Robustly extracts the full abstract from XML.
Handles multiple <AbstractText> sections and inline formatting tags (e.g., <i>).
It preserves the section labels (e.g., BACKGROUND, METHODS) for better context.
Args:
article_xml_element (xml.etree.ElementTree.Element): The root XML element for the article.
Returns:
A single string containing the full, cleaned abstract, or an empty string if not found.
"""
if not isinstance(article_xml_element, ET.Element):
return ""
abstract_element = article_xml_element.find('.//Abstract')
if abstract_element is None:
return ""
abstract_parts = []
# Iterate through all <AbstractText> nodes within the <Abstract> tag
for abstract_text_node in abstract_element.findall('AbstractText'):
# Get the section label (e.g., "BACKGROUND") if it exists
label = abstract_text_node.get('Label', '').strip()
# .itertext() correctly extracts all text, including from child tags like <i>
text_content = "".join(abstract_text_node.itertext()).strip()
if label:
# Format with the label for better readability and context for the LLM
abstract_parts.append(f"{label.upper()}: {text_content}")
else:
# If there's no label, just append the text
abstract_parts.append(text_content)
# Join all parts with a double newline to separate the sections
return "\n\n".join(abstract_parts)
</code>
<code>
# Initialize classifier
classifier = DOIClassifier(GEMINI_API_KEY, ICD11_CLIENT_ID, ICD11_CLIENT_SECRET)
# Process DOIs
doi_file = "dois.txt" # Change this to your DOI file path
output_file = "article_classification.csv"
results_df = classifier.process_dois(doi_file, output_file)
print(f"Processing complete! Found information for {len(results_df)} articles.")
</code>
<code>
results_df
</code>
<code>
results_df.iloc[6]
</code>
<code>
cl2.search_icd11_code("Small intestinal bacterial overgrowth")
</code>
<code>
cl2 = DOIClassifier(GEMINI_API_KEY, ICD11_CLIENT_ID, ICD11_CLIENT_SECRET)
cl2.classify_disease_with_gemini("Effects of Proton Pump Inhibitors on the Small Bowel and Stool Microbiomes.","""BACKGROUND: Proton pump inhibitor (PPI) use is extremely common. PPIs have been suggested to affect the gut microbiome, and increase risks of Clostridium difficile infection and small intestinal bacterial overgrowth (SIBO). However, existing data are based on stool analyses and PPIs act on the foregut.
AIMS: To compare the duodenal and stool microbiomes in PPI and non-PPI users.
METHODS: Consecutive subjects presenting for upper endoscopy without colonoscopy were recruited. Current antibiotic users were excluded. Subjects taking PPI were age- and gender-matched 1:2 to non-PPI controls. Subjects completed medical history questionnaires, and duodenal aspirates were collected using a validated protected catheter. A subset also provided stool samples. Duodenal and stool microbiomes were analyzed by 16S rRNA sequencing.
RESULTS: The duodenal microbiome exhibited no phylum-level differences between PPI (N = 59) and non-PPI subjects (N = 118), but demonstrated significantly higher relative abundances of families Campylobacteraceae (3.13-fold, FDR P value < 0.01) and Bifidobacteriaceae (2.9-fold, FDR P value < 0.01), and lower relative abundance of Clostridiaceae (88.24-fold, FDR P value < 0.0001), in PPI subjects. SIBO rates were not significantly different between groups, whether defined by culture (> 103 CFU/ml) or 16S sequencing, nor between subjects taking different PPIs. The stool microbiome exhibited significantly higher abundance of family Streptococcaceae (2.14-fold, P = 0.003), and lower Clostridiaceae (2.60-fold, FDR P value = 8.61E-13), in PPI (N = 22) versus non-PPI (N = 47) subjects.
CONCLUSIONS: These findings suggest that PPI use is not associated with higher rates of SIBO. Relative abundance of Clostridiaceae was reduced in both the duodenal and stool microbiomes, and Streptococcaceae was increased in stool. The clinical implications of these findings are unknown.""")
</code>
|
{
"filename": "Get_ICD_11.ipynb",
"repository": "dkisselev-zz/mmc-pipeline",
"query": "transformed_from_existing",
"size": 107876,
"sha": ""
}
|
# Slicing.ipynb
Repository: sagar87/spatialproteomics
# Subselecting Data
<code>
%reload_ext autoreload
%autoreload 2
import spatialproteomics
import pandas as pd
import xarray as xr
xr.set_options(display_style="text")
</code>
One of the key features of `spatialproteomics` is the ability to slice our image data quickly and intuitively. We start by loading our _spatialproteomics_ object.
<code>
ds = xr.open_zarr("../../data/BNHL_166_4_I2_LK_2.zarr")
</code>
## Slicing Channels and Spatial Coordinates
To slice specific channels of the image we simply use `.pp` accessor together with the familiar bracket `[]` indexing.
<code>
ds.pp["CD4"]
</code>
We can also select multiple channels by simply passing a list to the `.pp` accessor. As we will see later, this makes visualising image overlays easy.
<code>
ds.pp[["CD4", "CD8"]]
</code>
The `.pp` accessor also understands `x` and `y` coordinates. When `x` and `y` coordinates are sliced, we get ridd of all cells that do not belong to the respective image slice.
<code>
ds.pp[50:150, 50:150]
</code>
Note that we can also pass `channels` and `x, y` coordinates at the same time.
<code>
ds.pp[["CD4", "CD8"], 50:150, 50:150]
</code>
## Slicing Labels
The labels accessor `.la` allows to select specific cell types by their label number or name.
<code>
ds.la[4]
</code>
<code>
ds.la["T (CD3)"]
</code>
Again it is possible to pass multiple cell labels.
<code>
ds.la[4, 5, 6]
</code>
Finally, we can select all cells except a cell type using `la.deselect`.
<code>
ds.la.deselect([1])
</code>
## Slicing Neighborhoods
We can also select by neighborhoods with the `nh` accessor. The syntax is identical to the one in the label subsetting.
<code>
ds = xr.open_zarr("../../data/sample_1_with_neighborhoods.zarr")
ds
</code>
<code>
# subsetting only neighborhood 0
ds.nh[0]
</code>
|
{
"filename": "Slicing.ipynb",
"repository": "sagar87/spatialproteomics",
"query": "transformed_from_existing",
"size": 39343,
"sha": ""
}
|
# Make_a_chatbot_from_scratch.ipynb
Repository: insaid2018/project-gallery
<center><img src="https://github.com/insaid2018/Term-1/blob/master/Images/INSAID_Full%20Logo.png?raw=true" width="25%" /></center>
# <center><b>Making of a simple interactive chatbot<b></center>
# **Table of Contents**
---
**1.** [**Problem Statement**](#Section1)<br>
**2.** [**Importing Libraries**](#Section2)<br>
**3.** [**Data-Corpus Formation**](#Section3)<br>
**4.** [**Preprocessing**](#Section4)<br>
**5.** [**Greetings**](#Section5)<br>
**6.** [**Genarate response**](#Section6)<br>
**7.** [**Applications**](#Section7)<br>
**8.** [**Limitations**](#Section8)<br>
**9.** [**Conclusion**](#Section9)<br>
---
<a name = Section1></a>
# **1. Problem Statement**
---
- A **chatbot or chatterbot** is a **software application** used to **conduct an on-line chat conversation** via **text or text-to-speech**, in lieu of providing direct contact with a live human agent
- The goal of this project is to develop a **simple chatbot** from the scratch.
<br>
<center><img src="https://trymondo.com/wp-content/uploads/2020/11/Chatbot.gif" /></center>
### **Scenario:**
- You are working for a technical start up who are willing to launch their own chatbot.
- They want you to develop this **in house chatbot**.
- The bot should be able to return responses for a given set of input(s).
---
<a name = Section2></a>
# **2. Importing Libraries**
---
<code>
import nltk # importign nltk
from nltk.stem import WordNetLemmatizer # Importing wordnetlematizer
lemmatizer = WordNetLemmatizer() # Initiating instance
import json # importing json
import pickle # importing pickle
import numpy as np # importing numpy
from keras.models import Sequential # importing sequential layers
from keras.layers import Dense, Activation, Dropout # importing other layers
from tensorflow.keras.optimizers import SGD # importing SGD
import random # importing random
import tensorflow # importing tensorflow
from tensorflow.keras import Sequential # importing sequential
from tensorflow.keras.layers import Dense, Dropout # importing dense and dropout layers
nltk.download('punkt') # downloading punkt
nltk.download('wordnet') # downloading wordnet
import string # importing string
from sklearn.feature_extraction.text import TfidfVectorizer # importing TfIDF
from sklearn.metrics.pairwise import cosine_similarity # importing cosine similarity
</code>
---
<a name = Section3></a>
# **3. Data-Corpus Formation**
---
- Here, we will taking the .txt file which has been taken from <a href="https://en.wikipedia.org/wiki/Chatbot">here</a>
and make the **data corpus** out of it.
- A **data corpus** is a collection of linguistic data.
- Then we will set all the **sent tokens** and the **word tokens**.
<code>
# Reading the data from the json file
data = open('/content/AI Content for chatbot.txt','r', errors ='ignore')
raw = data.read()
# Making a list for all the words
all_words=[]
# Making a list for all the classes
all_classes = []
# Making a list for all documents
all_documents = []
# Ignoring ? and !
ignore_words = ['?', '!']
</code>
<code>
# Lowercasing every word of the corpus
raw = raw.lower()
</code>
<code>
# Raw data to sentence
sent_tokens = nltk.sent_tokenize(raw)
</code>
<code>
sent_tokens
</code>
<code>
# Raw data to list of words
word_tokens = nltk.word_tokenize(raw)
</code>
<code>
word_tokens
</code>
---
<a name = Section4></a>
# **4. Preprocessing**
---
<code>
# Creating a lemitization object
lemmer = nltk.stem.WordNetLemmatizer()
</code>
<code>
def Lemtokens(tokens):
return [lemmer.lemmatize(token) for token in tokens]
</code>
<code>
# removing punctutations:
remove_punc = dict((ord(x), None) for x in string.punctuation)
</code>
<code>
def LemNormalize(text):
return Lemtokens(nltk.word_tokenize(text.lower().translate(remove_punc)))
</code>
---
<a name = Section5></a>
# **5. Greetings**
---
- In this section we will making the greeting function.
- This greeting function will take a user input such as **Hello**.
- After this this greeting function will return the value of the corresponding greeting message such as **Hello there!**
- This greeting function has been made with only few of the elements which are there in the **`greeting_input_list`** and hence, the response is also limied.
<code>
greeting_input_list = ["hello","hi","greetings","what's up","hey"]
</code>
<code>
greeting_response_list = ['Hello there!', 'how are you', 'greetings to you too','hello!']
</code>
<code>
# Once called this will return response for every user input given:
def greet(sentence):
for word in sentence.split():
if word.lower() in greeting_input_list:
return(random.choice(greeting_response_list))
</code>
---
<a name = Section6></a>
# **6. Genarating Responses**
---
- In this section we will be feeding a list of inputs to the bot and genarate the responses from the bot accordingly.
- If the bot fails to predict the output of a response or the response is not known the bot will return **'Apologies! Could not understand that'**
<code>
def responses(user_input):
bot_responses = ''
sent_tokens.append(user_input)
vect = TfidfVectorizer(tokenizer=LemNormalize,
stop_words = 'english')
tfidf = vect.fit_transform(sent_tokens)
vals = cosine_similarity(tfidf[-1],
tfidf)
idx = vals.argsort()[0][-2]
flat = vals.flatten()
flat.sort()
req_tfidf = flat[-2]
if(req_tfidf==0):
bot_responses = bot_responses + 'Aplogies! Could not understand that'
return bot_responses
else:
bot_responses = bot_responses+sent_tokens(idx)
return bot_responses
</code>
<code>
flag = True
print('MyBOT: Hello there! I am the MyBOT and I am there to help you out and if you wanna quit simply type thank you or Bye')
while(flag == True):
user_input = input()
user_input = user_input.lower()
# print(user_input)
if (user_input == 'Bye' or user_input == 'bye'):
flag = False
print('MyBOT: It was great talking to you. Goodbye')
elif (user_input != 'Bye'):
if(user_input == 'thanks' or user_input == 'thank you'):
flag = False
print('MyBOT: Welcome')
else:
if(greet(user_input)!= None):
print('MyBOT: '+ greet(user_input))
else:
print('MyBOT: ',end='')
print(responses(user_input))
sent_tokens.remove(user_input)
</code>
---
<a name = Section7></a>
# **7. Applications**
---
- This chatbot if trained with a large amount of data can be used as a **general purpose chatbot**.
- Areas such as **BFSI, Retail and Ecommerce** will have a wide range of applications at the time of customer dealing with the help of this chatbot.
---
<a name = Section8></a>
# **8. Limitations**
---
- As because this is a **prototype** this will thorow errors as a bulk amount of data processing is required.
- The **accuracy** of this bot is relatively low as because of **the size of the training data corpus**.
---
<a name = Section9></a>
# **9. Conclusion**
---
- In this project we have successfully developed a chatbot that can deal with **human responses.**
- This can be further tuned to develop a **human handoff** such as **RASA NLU**
- As because this is a prototype and the size of the **training data corpus** this chatbot is throwing with low accuracy and can accurately work the data that has been fed.
|
{
"filename": "Make_a_chatbot_from_scratch.ipynb",
"repository": "insaid2018/project-gallery",
"query": "transformed_from_existing",
"size": 107147,
"sha": ""
}
|
# feature_selection.ipynb
Repository: shumshersubashgautam/Single-Cell-Mapping-Computational-Biology
(pre-processing:feature-selection)=
# Feature selection
## Motivation
We now have a normalized data representation that still preserves biological heterogeneity but with reduced technical sampling effects in gene expression. Single-cell RNA-seq datasets usually contain up to 30,000 genes and so far we only removed genes that are not detected in at least 20 cells. However, many of the remaining genes are not informative and contain mostly zero counts. Therefore, a standard preprocessing pipeline involves the step of feature selection which aims to exclude uninformative genes which might not represent meaningful biological variation across samples.
:::{figure-md} Feature selection
<img src="../_static/images/preprocessing_visualization/feature_selection.jpeg" alt="Feature selection" class="bg-primary mb-1" width="800px">
Feature selection generally describes the process of only selecting a subset of relevant features which can be the most informative, most variable or most deviant ones.
:::
Usually, the scRNA-seq experiment and resulting dataset focuses on one specific tissue and hence, only a small fraction of genes is informative and biologically variable. Traditional approaches and pipelines either compute the coefficient of variation (highly variable genes) or the average expression level (highly expressed genes) of all genes to obtain 500-2000 selected genes and use these features for their downstream analysis steps. However, these methods are highly sensitive to the normalization technique used before. As mentioned earlier, a former preprocessing workflow included normalization with CPM and subsequent log transformation. But as log-transformation is not possible for exact zeros, analysts often add a small *pseudo count*, e.g., 1 (log1p), to all normalized counts before log transforming the data. Choosing the pseudo count, however, is arbitrary and can introduce biases to the transformed data. This arbitrariness has then also an effect on the feature selection as the observed variability depends on the chosen pseudo count. A small pseudo count value close to zero is increasing the variance of genes with zero counts {cite}`Townes2019`.
Germain et al. instead proposes to use *deviance* for feature selection which works on raw counts {cite}`fs:germain_pipecomp_2020`. Deviance can be computed in closed form and quantifies whether genes show a constant expression profile across cells as these are not informative. Genes with constant expression are described by a multinomial null model, they are approximated by the binomial deviance. Highly informative genes across cells will have a high deviance value which indicates a poor fit by the null model (i.e., they don't show constant expression across cells). According to the deviance values, the method then ranks all genes and obtains only highly deviant genes.
As mentioned before, deviance can be computed in closed form and is provided within the R package scry.
We start by setting up our environment.
<code>
import scanpy as sc
import anndata2ri
import logging
import matplotlib.pyplot as plt
import numpy as np
import seaborn as sns
import rpy2.rinterface_lib.callbacks as rcb
import rpy2.robjects as ro
sc.settings.verbosity = 0
sc.settings.set_figure_params(
dpi=80,
facecolor="white",
frameon=False,
)
rcb.logger.setLevel(logging.ERROR)
ro.pandas2ri.activate()
anndata2ri.activate()
%load_ext rpy2.ipython
</code>
<code>
%%R
library(scry)
</code>
Next, we load the already normalized dataset. Deviance works on raw counts so there is no need to replace `adata.X` with one of the normalized layers, but we can directly use the object as it was stored in the normalization notebook.
<code>
adata = sc.read(
filename="s4d8_normalization.h5ad",
backup_url="https://figshare.com/ndownloader/files/40015741",
)
</code>
Similar to before, we save the AnnData object in our R environment.
<code>
ro.globalenv["adata"] = adata
</code>
We can now directly call feature selection with deviance on the non-normalized counts matrix and export the bionomial deviance values as a vector.
<code>
%%R
sce = devianceFeatureSelection(adata, assay="X")
</code>
<code>
binomial_deviance = ro.r("rowData(sce)$binomial_deviance").T
</code>
As a next step, we now sort the vector an select the top 4,000 highly deviant genes and save them as an additional column in `.var` as 'highly_deviant'. We additionally save the computed binomial deviance in case we want to sub-select a different number of highly variable genes afterwards.
<code>
idx = binomial_deviance.argsort()[-4000:]
mask = np.zeros(adata.var_names.shape, dtype=bool)
mask[idx] = True
adata.var["highly_deviant"] = mask
adata.var["binomial_deviance"] = binomial_deviance
</code>
Last, we visualise the feature selection results. We use a scanpy function to compute the mean and dispersion for each gene accross all cells.
<code>
sc.pp.highly_variable_genes(adata, layer="scran_normalization")
</code>
We inspect our results by plotting dispersion versus mean for the genes and color by 'highly_deviant'.
<code>
ax = sns.scatterplot(
data=adata.var, x="means", y="dispersions", hue="highly_deviant", s=5
)
ax.set_xlim(None, 1.5)
ax.set_ylim(None, 3)
plt.show()
</code>
We observe that genes with a high mean expression are selected as highly deviant. This is in agreement with emprical observations by {cite}`Townes2019`.
<code>
adata.write("s4d8_feature_selection.h5ad")
</code>
## References
```{bibliography}
:filter: docname in docnames
:labelprefix: fs
```
## Contributors
We gratefully acknowledge the contributions of:
### Authors
* Anna Schaar
### Reviewers
* Lukas Heumos
|
{
"filename": "feature_selection.ipynb",
"repository": "shumshersubashgautam/Single-Cell-Mapping-Computational-Biology",
"query": "transformed_from_existing",
"size": 144632,
"sha": ""
}
|
# T5_1.ipynb
Repository: LaurentVeyssier/Abstractive-Summarization-using-colab-and-T5-model
<code>
!pip install transformers
!pip install tensorflow==2.1
from transformers import pipeline
</code>
<code>
summarizer = pipeline("summarization", model="t5-base", tokenizer="t5-base", framework="tf")
summarizer("The US has passed the peak on new coronavirus cases, President Donald Trump said and predicted that some states would reopen this month. The US has over 637,000 confirmed Covid-19 cases and over 30,826 deaths, the highest for any country in the world. At the daily White House coronavirus briefing on Wednesday, Trump said new guidelines to reopen the country would be announced on Thursday after he speaks to governors. We'll be the comeback kids, all of us, he said. We want to get our country back. The Trump administration has previously fixed May 1 as a possible date to reopen the world's largest economy, but the president said some states may be able to return to normalcy earlier than that.", min_length=25, max_length=100)
</code>
<code>
summarizer("I do not have friends in tennis, says Maria Sharapova. Maria Sharapova has basically no friends as tennis players on the WTA Tour. The Russian player has no problems in openly speaking about it and in a recent interview she said: I don't really hide any feelings too much. I think everyone knows this is my job here. When I'm on the courts or when I'm on the court playing, I'm a competitor and I want to beat every single person whether they're in the locker room or across the net. So I'm not the one to strike up a conversation about the weather and know that in the next few minutes I have to go and try to win a tennis match. I'm a pretty competitive girl. I say my hellos, but I'm not sending any players flowers as well. Uhm, I'm not really friendly or close to many players. I have not a lot of friends away from the courts. When she said she is not really close to a lot of players, is that something strategic that she is doing? Is it different on the men's tour than the women's tour? No, not at all. I think just because you're in the same sport doesn't mean that you have to be friends with everyone just because you're categorized, you're a tennis player, so you're going to get along with tennis players. I think every person has different interests. I have friends that have completely different jobs and interests, and I've met them in very different parts of my life. I think everyone just thinks because we're tennis players we should be the greatest of friends. But ultimately tennis is just a very small part of what we do. There are so many other things that we're interested in, that we do.", min_length=25, max_length=100)
</code>
<code>
text="The COVID-19 pandemic is disrupting the daily lives of people all around the world. But what about the ways they stay connected with loved ones? Richard Slatcher, the Gail M. Williamson Distinguished Professor of Psychology at the University of Georgia, is working with two international colleagues to determine the psychological effects of a decrease in face-to-face communication with their project. The COVID-19 outbreak is profoundly affecting our social relationships. Are people feeling more or less connected to others? How are couples feeling about working from home together? What are the effects of people working full time from home while also caring full time for their children? What are the effects of living alone right now? said Slatcher, whose research focuses on how people’s relationships with others can affect their well-being and health. This experience will impact us in ways we don’t yet fully understand. The researchers are gathering data through a survey, hoping to connect with as many people as possible from around the world and hear stories of how the pandemic is altering their relationships and well-being, Slatcher said. With this information, the researchers will gauge how the pandemic affects people from different countries and cultures. This study is really about relationships: how the pandemic is influencing how connected people feel to others, Slatcher said. Many people will feel very isolated, both physically and psychologically, but others may actually feel more connected to their households, neighbors and/or social networks. In fact, since launching our study, we have already heard from some people reporting that they feel more connected to others than they typically do. The way people are connecting during this time is incredibly moving—and not despite the pandemic, but because of it, Zoppolat said. We are inherently social beings, and this deep drive for connection becomes beautifully and painfully apparent in times like these. The research could help scientists understand which types of people are the most psychologically vulnerable to the pandemic’s effects by finding predictors of who will struggle the most with isolation."
</code>
<code>
summarizer(text, min_length=25, max_length=100)
</code>
<code>
text="en 1894, le capitaine de l'armée française Alfred Dreyfus, Juif, accusé d'avoir livré aux Allemands des documents secrets, est condamné au bagne à perpétuité pour trahison et déporté. À cette date, l'opinion comme la classe politique française est unanimement défavorable à Dreyfus. Certaine de l'incohérence de cette condamnation, la famille du capitaine, derrière son frère Mathieu, tente de prouver son innocence, engageant à cette fin le journaliste Bernard Lazare. Parallèlement, le colonel Georges Picquart, chef du contre-espionnage, constate en mars 1896 que le vrai traître est le commandant Ferdinand Walsin Esterhazy. L'état-major refuse pourtant de revenir sur son jugement et affecte Picquart en Afrique du Nord. Afin d'attirer l'attention sur la fragilité des preuves contre Dreyfus, sa famille décide de contacter en juillet 1897 le respecté vice-président du Sénat Auguste Scheurer-Kestner qui fait savoir, trois mois plus tard, qu'il a acquis la conviction de l'innocence de Dreyfus, et qui en persuade également Georges Clemenceau, ancien député et alors simple journaliste. Le même mois, Mathieu Dreyfus porte plainte auprès du ministère de la Guerre contre Walsin Esterhazy. Alors que le cercle des dreyfusards s'élargit, deux événements quasi simultanés donnent en janvier 1898 une dimension nationale à l'affaire : Esterhazy est acquitté sous les acclamations des conservateurs et des nationalistes ; Émile Zola publie « J'accuse… ! », réquisitoire dreyfusard qui entraîne le ralliement de nombreux intellectuels. Un processus de scission de la France est entamé, et se prolonge jusqu'à la fin du siècle."
</code>
<code>
summarizer(text, min_length=25, max_length=100)
</code>
<code>
text="Lebanese officials targeted in the investigation of the massive blast that tore through Beirut sought to shift blame for the presence of explosives at the city’s port, and the visiting French president warned Thursday that without serious reforms the country would “continue to sink.” The blast Tuesday, which appeared to have been caused by an accidental fire that ignited a warehouse full of ammonium nitrate at the city’s port, rippled across the Lebanese capital, killing at least 135 people, injuring more than 5,000 and causing widespread destruction. It also may have accelerated the country’s coronavirus outbreak, as thousands flooded into hospitals in the wake of the blast. Tens of thousands have been forced to move in with relatives and friends after their homes were damaged, further raising the risks of exposure. French President Emmanuel Macron visited Thursday amid widespread pledges of international aid. It’s unclear how much support the international community will offer the notoriously corrupt and dysfunctional government. Macron, who viewed the devastated port and was to meet with senior Lebanese officials, said the visit is “an opportunity to have a frank and challenging dialogue with the Lebanese political powers and institutions.”,He said France will work to coordinate aid but warned that “if reforms are not made, Lebanon will continue to sink.”,Later, as he toured one of the hardest-hit neighborhoods, an angry crowd vented its fury at Lebanon’s political leaders, chanting “Revolution” and “The people want to bring down the regime,” slogans used during mass protests last year.,Macron said he was not there to endorse the “regime” and vowed that French aid would not fall into the “hands of corruption.” Losses from the blast are estimated to be between $10 billion to $15 billion, Beirut Gov. Marwan Abboud told the Saudi-owned TV station Al-Hadath on Wednesday, adding that nearly 300,000 people are homeless. The head of Lebanon’s customs department meanwhile confirmed in an interview with LBC TV late Wednesday that officials had sent five or six letters over the years to the judiciary asking that the ammonium nitrate be removed because of the dangers it posed."
</code>
<code>
summarizer(text, min_length=25, max_length=100)
</code>
<code>
text="It is no secret to the campaigns of Joe Biden and Donald Trump that the road to the White House runs through places like Michigan’s Macomb County. It is a swing county in one of a trio of recently reliably Democratic states – Michigan, Pennsylvania, Wisconsin – that shocked Hillary Clinton’s 2016 campaign by breaking for Trump after backing Barack Obama in 2008 and 2012. The county, a suburban and exurban area north of Detroit, is the state’s third-most populous. Eighty percent of its residents are white. Roughly a quarter of adults have college degrees. The median household income in 2018 was about $60,000. Voters there cast ballots at higher rates than the country overall. It is a bellwether that backed the candidate elected president all but three times in the past 50 years. Simply put, Macomb County is chock-full of people whose demographic and political profiles make them highly sought-after by political strategists from both parties in Washington as potentially persuadable voters. In the 2020 White House race, top polls show that Biden is widening his lead over Trump in Michigan. The president’s reelection campaign has stopped buying television and radio ads in the state, and studies indicate that white suburban and working-class women are more likely to be having second thoughts about Trump than their male counterparts. Nevertheless, interviews late last month with nearly two dozen Macomb County women fitting this profile show that right now, in this swing county in this swing state, neither candidate has a lock. As Kristina Gallagher, a 36-year-old married mother of two, put it: “I’m a realist. I’m going to vote for the person who is going to do the best for us.” Gallagher is neither a Republican nor a Democrat, voting for Obama in 2008, Republican Mitt Romney in 2012 and Trump in 2016. She is a waitress who, for the first time in a decade, has a little money in the bank and wants a president focused on jobs, health insurance and education. Will she vote for Trump again? “He’s been a little iffy lately, can’t keep his mouth shut. He’s derogatory sometimes and it really irks me. Maybe stay off Twitter,” Gallagher said as she loaded groceries, and her daughter, into the car outside a Walmart in Roseville. What about Biden? “Not sure,” Gallagher said."
</code>
<code>
summarizer(text, min_length=25, max_length=100)
</code>
|
{
"filename": "T5_1.ipynb",
"repository": "LaurentVeyssier/Abstractive-Summarization-using-colab-and-T5-model",
"query": "transformed_from_existing",
"size": 72928,
"sha": ""
}
|
# Pubmed.ipynb
Repository: Aitslab/BioNLP
<code>
from docria import Document, DataTypes as T, NodeSpan, set_large_screen, MsgpackCodec, MsgpackDocument
from docria.storage import MsgpackDocumentIO, MsgpackDocumentReader, MsgpackDocumentWriter
from lxml import etree
import regex as re
</code>
## Import
<code>
%%sh
zcat pubmed_mini/pubmed19n0080.xml.gz | head -n 100
</code>
<code>
pubmed0080 = etree.parse("pubmed_mini/pubmed19n0080.xml.gz")
</code>
<code>
articles = pubmed0080.iterfind("PubmedArticle")
</code>
<code>
article = next(articles)
</code>
<code>
print(etree.tounicode(article))
</code>
<code>
article.find(".//Article/ArticleTitle").text
</code>
<code>
article.find(".//PMID").text
</code>
<code>
abstract = article.find(".//Abstract")
</code>
<code>
print(etree.tounicode(abstract))
</code>
<code>
descedants = list(abstract.iterdescendants())
</code>
<code>
descedants
</code>
<code>
abstract_text = "".join(node.text for node in descedants if node.text is not None).strip()
print(abstract_text)
</code>
## Processing prototyping
Test the regex at https://www.regex101.com
<code>
last = 0
for m in re.finditer(r"(?>[\.\?\!])(?:\s*(?=\p{Lu})|$)", abstract_text):
print("SENT:", abstract_text[last:m.start()+1])
last = m.end()
</code>
<code>
set_large_screen()
</code>
<code>
doc = Document()
</code>
<code>
doc.props["pmid"] = article.find(".//PMID").text
</code>
<code>
doc.maintext = abstract_text
</code>
<code>
doc
</code>
<code>
doc.add_layer("token", text=T.span(), partOfSpeech=T.string, namedEntity=T.string("O"), indx=T.int32)
doc.add_layer("sentence", text=T.span(), tokens=T.nodespan("token"))
</code>
<code>
sentence_layer = doc["sentence"]
token_layer = doc["token"]
last = 0
for m in re.finditer(r"(?>[\.\?\!])(?:\s*(?=\p{Lu})|$)", abstract_text):
tokens = []
text = doc.maintext[last:m.start()+1]
offset = last
for tok_i, tok_m in enumerate(re.finditer(r"\p{L}+|\p{N}+(\.\p{N}+)?|[\-\/%():,\.;+&#=!?@_<>]", str(text))):
tokens.append(token_layer.add(indx=tok_i+1, text=doc.maintext[tok_m.start()+offset:tok_m.end()+offset]))
if len(tokens) > 0:
sentence = sentence_layer.add(text=text)
sentence["tokens"] = NodeSpan(tokens[0], tokens[-1])
last = m.end()
</code>
<code>
doc["sentence"]
</code>
<code>
doc["sentence"]["tokens"][0]
</code>
<code>
doc["token"][doc["token"]["text"] == "of"]
</code>
<code>
from docria.algorithm import group_by_span
</code>
<code>
group_by_span?
</code>
Hint: The statement below works because, 'text' is the default name of a span, if it is not called 'text', either group_span_field or layer_span_field = {'token': name} must be set.
<code>
of_tokens = doc["token"][doc["token"]["text"] == "of"]
</code>
<code>
group_by_span(group_nodes=doc["sentence"], layer_nodes={"token": of_tokens})
</code>
## Scale it up, processing many documents
<code>
def segment(doc):
token_layer = doc.add_layer("token", text=T.span(), partOfSpeech=T.string, namedEntity=T.string("O"), indx=T.int32)
sentence_layer = doc.add_layer("sentence", text=T.span(), tokens=T.nodespan("token"))
pubmed_abstract = doc.texts["main"]
sentence_layer = doc["sentence"]
token_layer = doc["token"]
last = 0
for m in re.finditer(r"(?>[\.\?\!])(?:\s*(?=\p{Lu})|$)", str(pubmed_abstract)):
tokens = []
text = pubmed_abstract[last:m.start()+1]
offset = last
for tok_i, tok_m in enumerate(re.finditer(r"\p{L}+|\p{N}+(\.\p{N}+)?|[\-\/%():,\.;+&#=!?@_<>]", str(text))):
tokens.append(token_layer.add(indx=tok_i+1, text=doc.maintext[tok_m.start()+offset:tok_m.end()+offset]))
if len(tokens) > 0:
sentence = sentence_layer.add(text=text)
sentence["tokens"] = NodeSpan(tokens[0], tokens[-1])
last = m.end()
</code>
<code>
def process_pubmed(articles):
for article in articles:
title = article.find(".//Article/ArticleTitle").text
pmid = article.find(".//PMID").text
abstract = article.find(".//Abstract")
if abstract is None:
continue
abstract_text = "".join(node.text for node in abstract.iterdescendants() if node.text is not None).strip()
assert pmid is not None
assert title is not None
assert abstract is not None
doc = Document()
doc.props["pmid"] = pmid
doc.props["title"] = title
doc.maintext = abstract_text
segment(doc)
yield doc
</code>
<code>
def process(inputfile, outputfile):
pubmedxml = etree.parse(inputfile)
with open(outputfile, "wb") as fout, MsgpackDocumentWriter(fout) as writer:
for doc in process_pubmed(pubmedxml.iterfind("PubmedArticle")):
writer.write(doc)
</code>
<code>
process("pubmed_mini/pubmed19n0080.xml.gz", "pubmed00080.docria")
</code>
<code>
from tqdm import tqdm
</code>
<code>
reader = MsgpackDocumentReader(open("pubmed00080.docria", "rb"))
</code>
<code>
pmids = []
titles = []
</code>
<code>
for doc in tqdm(reader):
props = doc.properties()
pmids.append(props["pmid"])
titles.append(props["title"])
</code>
<code>
pmids[0:100]
</code>
<code>
titles[0:100]
</code>
<code>
sentences = []
</code>
<code>
reader = MsgpackDocumentReader(open("pubmed00080.docria", "rb"))
for mdoc in tqdm(reader):
doc = mdoc.document()
sentences.extend([str(sent["text"]) for sent in doc["sentence"]])
</code>
<code>
sentences[0:100]
</code>
<code>
from multiprocessing import Pool, cpu_count
</code>
<code>
import os
</code>
<code>
inputfiles = list(
map(lambda fname: os.path.join("pubmed_mini", fname),
filter(lambda fname: fname.endswith(".xml.gz"), os.listdir("pubmed_mini"))
)
)
</code>
<code>
inputfiles
</code>
<code>
outputfiles = [os.path.join("pubmed_mini", os.path.basename(fname) + ".docria") for fname in inputfiles]
</code>
<code>
outputfiles
</code>
<code>
def genwork(inputfiles, outputfiles):
for i, o in zip(inputfiles, outputfiles):
yield {"inputfile": i , "outputfile": o}
</code>
<code>
def work(args):
inputfile = args["inputfile"]
outputfile = args["outputfile"]
process(inputfile, outputfile)
return outputfile
</code>
<code>
pool = Pool(cpu_count())
</code>
<code>
pool.imap_unordered?
</code>
<code>
for completed in tqdm(pool.imap_unordered(work, genwork(inputfiles, outputfiles))):
pass
</code>
|
{
"filename": "Pubmed.ipynb",
"repository": "Aitslab/BioNLP",
"query": "transformed_from_existing",
"size": 100709,
"sha": ""
}
|
# EnrichrConsensus_1.ipynb
Repository: MaayanLab/appyter-catalog
<code>
#%%appyter init
from appyter import magic
magic.init(lambda _=globals: _())
</code>
<code>
%%appyter hide_code
{% do SectionField(
name='PRIMARY',
title='Enrichr Consensus Terms',
subtitle='This appyter returns consensus Enrichr terms using a set of gene sets',
img='enrichr.png'
) %}
</code>
<code>
%%appyter code_exec
{% set title = StringField(
name='title',
label='Notebook name',
default='Enrichr Consensus Terms',
section="PRIMARY",
) %}
title = {{ title }}
</code>
<code>
import time
import requests
import pandas as pd
import json
import seaborn as sns
import matplotlib.pyplot as plt
from IPython.display import display, IFrame, Markdown
import math
import scipy.stats as st
import fastcluster
</code>
<code>
display(Markdown("# %s"%(title)), display_id="title")
</code>
<code>
clustergrammer_url = 'https://amp.pharm.mssm.edu/clustergrammer/matrix_upload/'
ENRICHR_URL = 'https://maayanlab.cloud/Enrichr'
# libraries = ["ChEA_2016", "GO_Biological_Process_2018" ,"GWAS_Catalog_2019" , "KEGG_2019_Human"]
table = 1
figure = 1
</code>
<code>
def clustergrammer(df, name, clustergrammer_url, display_id, fignum=1, label="Clustergrammer"):
clustergram_df = df.rename(columns={i:"Signature: %s"%i for i in df.columns}, index={i:"Enriched Term: %s"%i for i in df.index})
clustergram_df.to_csv(name, sep="\t")
response = ''
for i in range(5):
try:
res = requests.post(clustergrammer_url, files={'file': open(name, 'rb')})
if not res.ok:
response = res.text
time.sleep(1)
else:
clustergrammer_url = res.text.replace("http:","https:")
break
except Exception as e:
response = e
time.sleep(2)
else:
if type(response) == Exception:
raise response
else:
raise Exception(response)
display(IFrame(clustergrammer_url, width="1000", height="1000"), display_id="clustergram_%s"%display_id)
display(Markdown("**Figure %d** %s [Go to url](%s)"%(fignum, label, clustergrammer_url)), display_id="clustergram_label_%s"%display_id )
</code>
<code>
cmap = sns.cubehelix_palette(50, hue=0.05, rot=0, light=1, dark=0)
def heatmap(df, filename, display_id, width=15, height=15):
# fig = plt.figure(figsize=(width,height))
cg = sns.clustermap(df, cmap=cmap, figsize=(width, height), cbar_pos=(0.02, 0.65, 0.05, 0.18),)
cg.ax_row_dendrogram.set_visible(False)
cg.ax_col_dendrogram.set_visible(False)
display(cg, display_id="heatmap_%s"%display_id)
plt.show()
cg.savefig(filename)
</code>
<code>
def get_dataframe(enrichment, lib, table, display_id):
term_df = pd.DataFrame(index=enrichment.keys())
for k,v in enrichment.items():
sigs = v["libraries"][lib]
for sig in sigs:
term = sig[1]
if term not in term_df.columns:
term_df[term] = 0.0
p = sig[2]
term_df.at[k, term] = -math.log(p)
term_df = term_df.transpose()
term_df.to_csv("%s_enrichment.tsv"%lib, sep="\t")
display(term_df.head(10), display_id="dataframe_%s"%display_id)
display(Markdown("**Table %d** The table below shows the result of the enrichment analysis of %d gene sets \
with the %s library in Enrichr. Each score is computed by getting the negative logarithm of the p-value \
($-\ln{pval}$). [Download complete table](%s_enrichment.tsv)"%(table, num_sigs, lib.replace("_"," "), lib)),
display_id="dataframe_caption_%s"%display_id
)
table+=1
return term_df, table
def get_consensus(df, lib, top_results, table, display_id):
consensus = df.sum(1).sort_values(ascending=False)[0:top_results].to_frame(name="scores")
# Save to tsv
consensus.to_csv("%s_consensus.tsv"%lib, sep="\t")
display(consensus.head(10), display_id="consensus_%s"%display_id)
display(Markdown("**Table %d** %s Consensus terms. \
Consensus scores are computed by taking the sum of scores in Table %d. \
[Download top %d terms](%s_consensus.tsv)"%(table, lib.replace("_"," "), (table-1), top_results, lib)),
display_id="consensus_caption_%s"%display_id
)
table+=1
return consensus, table
def stackedBarPlot(df, filename, display_id, width = 15, height = 15):
df['mean'] = df.mean(axis=1)
df = df.sort_values(by = 'mean')[0:top_results]\
.drop(['mean'], axis = 1)
if df.shape[0]==0:
return False
plot = df.plot.barh(stacked = True, figsize = (width,height), fontsize = 20)
plt.legend(bbox_to_anchor=(1.7, 0), loc='lower right', prop={'size': 16})
plt.xlabel('-log(p)',labelpad = 20, fontsize = 'xx-large')
display(plot, display_id="stacked_%s"%display_id)
plt.savefig(filename, format = 'svg', bbox_inches='tight')
plt.show()
return True
</code>
<code>
# Enrichr Functions
def addList(genes, description):
payload = {
'list': (None, '\n'.join(genes)),
'description': (None, description)
}
res = requests.post(ENRICHR_URL + "/addList", files=payload)
time.sleep(1)
if not res.ok:
raise Exception('Error analyzing gene list')
data = res.json()
return data["userListId"]
def enrich(userListId, library, alpha):
res = requests.get(
ENRICHR_URL +"/enrich", params={"userListId": userListId, "backgroundType": library}
)
time.sleep(1)
if not res.ok:
raise Exception('Error fetching enrichment results')
data = res.json()
return [i for i in data[library] if i[2] < alpha]
</code>
## Get Input
<code>
%%appyter code_exec
{% set input_gene_set = FileField(
name='input_gene_set',
label='Gene Set',
default='input.gmt',
section="PRIMARY",
examples={
'input.gmt': 'https://appyters.maayanlab.cloud/storage/EnrichrConsensus/sample_input/10input.gmt'
}
) %}
input_gene_set = {{ input_gene_set }}
</code>
<code>
%%appyter code_exec
transcription_libraries = {{ MultiChoiceField(name='transcription_libraries',
description='Select the Enrichr libraries you would like in your figure.',
label='Transcription',
default=[],
section = 'PRIMARY',
choices=[
'ARCHS4_TFs_Coexp',
'ChEA_2016',
'ENCODE_and_ChEA_Consensus_TFs_from_ChIP-X',
'ENCODE_Histone_Modifications_2015',
'ENCODE_TF_ChIP-seq_2015',
'Epigenomics_Roadmap_HM_ChIP-seq',
'Enrichr_Submissions_TF-Gene_Coocurrence',
'Genome_Browser_PWMs',
'lncHUB_lncRNA_Co-Expression',
'miRTarBase_2017',
'TargetScan_microRNA_2017',
'TF-LOF_Expression_from_GEO',
'TF_Perturbations_Followed_by_Expression',
'Transcription_Factor_PPIs',
'TRANSFAC_and_JASPAR_PWMs',
'TRRUST_Transcription_Factors_2019']) }}
pathways_libraries = {{ MultiChoiceField(name='pathways_libraries',
description='Select the Enrichr libraries you would like in your figure.',
label='Pathways',
default=[],
section = 'PRIMARY',
choices=[
'ARCHS4_Kinases_Coexp',
'BioCarta_2016',
'BioPlanet_2019',
'BioPlex_2017',
'CORUM',
'Elsevier_Pathway_Collection',
'HMS_LINCS_KinomeScan',
'HumanCyc_2016',
'huMAP',
'KEA_2015',
'KEGG_2019_Human',
'KEGG_2019_Mouse',
'Kinase_Perturbations_from_GEO_down',
'Kinase_Perturbations_from_GEO_up',
'L1000_Kinase_and_GPCR_Perturbations_down',
'L1000_Kinase_and_GPCR_Perturbations_up',
'NCI-Nature_2016',
'NURSA_Human_Endogenous_Complexome',
'Panther_2016',
'Phosphatase_Substrates_from_DEPOD',
'PPI_Hub_Proteins',
'Reactome_2016',
'SILAC_Phosphoproteomics',
'SubCell_BarCode',
'Virus-Host_PPI_P-HIPSTer_2020',
'WikiPathways_2019_Human',
'WikiPathways_2019_Mouse']) }}
ontologies_libraries = {{ MultiChoiceField(name='ontologies_libraries',
description='Select the Enrichr libraries you would like in your figure.',
label='Ontologies',
default=[],
section = 'PRIMARY',
choices=[
'GO_Biological_Process_2018',
'GO_Cellular_Component_2018',
'GO_Molecular_Function_2018',
'Human_Phenotype_Ontology',
'Jensen_COMPARTMENTS',
'Jensen_DISEASES',
'Jensen_TISSUES',
'MGI_Mammalian_Phenotype_Level_4_2019']) }}
diseases_drugs_libraries = {{ MultiChoiceField(name='diseases_drugs_libraries',
description='Select the Enrichr libraries you would like in your figure.',
label='Diseases/Drugs',
default=[],
section = 'PRIMARY',
choices=[
'Achilles_fitness_decrease',
'Achilles_fitness_increase',
'ARCHS4_IDG_Coexp',
'ClinVar_2019',
'dbGaP',
'DepMap_WG_CRISPR_Screens_Broad_CellLines_2019',
'DepMap_WG_CRISPR_Screens_Sanger_CellLines_2019',
'DisGeNET',
'DrugMatrix',
'DSigDB',
'GeneSigDB',
'GWAS_Catalog_2019',
'LINCS_L1000_Chem_Pert_down',
'LINCS_L1000_Chem_Pert_up',
'LINCS_L1000_Ligand_Perturbations_down',
'LINCS_L1000_Ligand_Perturbations_up',
'MSigDB_Computational',
'MSigDB_Oncogenic_Signatures',
'Old_CMAP_down',
'Old_CMAP_up',
'OMIM_Disease',
'OMIM_Expanded',
'PheWeb_2019',
'Rare_Diseases_AutoRIF_ARCHS4_Predictions',
'Rare_Diseases_AutoRIF_Gene_Lists',
'Rare_Diseases_GeneRIF_ARCHS4_Predictions',
'Rare_Diseases_GeneRIF_Gene_Lists',
'UK_Biobank_GWAS_v1',
'Virus_Perturbations_from_GEO_down',
'Virus_Perturbations_from_GEO_up',
'VirusMINT'])
}}
</code>
<code>
libraries = transcription_libraries + pathways_libraries + ontologies_libraries + diseases_drugs_libraries
</code>
<code>
enrichment = {}
with open(input_gene_set) as o:
for line in o:
unpacked = line.strip().split("\t")
if len(unpacked) == 1:
raise ValueError("Line '%s' is either empty or not formatted properly. Please consult README for more information"%line)
sigid = unpacked[0]
geneset = [i for i in unpacked[1:] if len(i) > 0]
enrichment[sigid] = {
"genes": [i.split(",")[0] for i in geneset]
}
</code>
<code>
num_sigs = len(enrichment)
input_sigs = pd.DataFrame.from_dict(enrichment, orient="index")
display(input_sigs.head(10))
display(Markdown("**Table %d** Input Signatures"%(table)), display_id="input_sigs")
table+=1
</code>
## User defined parameters
<code>
%%appyter code_exec
alpha = {{FloatField(name='alpha', label='p-value cutoff', default=0.05, section='PRIMARY')}}
top_results = {{IntField(name='min_count', label='Top results', description="Number of top results to keep", default=25, section='PRIMARY')}}
width = {{FloatField(name='width', label='image width', default=15, section='PRIMARY')}}
height = {{FloatField(name='height', label='image height', default=15, section='PRIMARY')}}
</code>
## Enrichment
<code>
failed_userlist = []
failed_enrich = {}
for description, values in enrichment.items():
print("Querying %s"%(description), end="\r", flush=True)
genes = values["genes"]
for tries in range(5):
try:
userListId = addList(genes, description)
enrichment[description]["userListId"] = userListId
break
except Exception as e:
print(e)
time.sleep(0.5)
else:
failed_userlist.append(description)
continue
time.sleep(0.1)
enrichment[description]["libraries"] = {}
for library in libraries:
for tries in range(5):
try:
userlistId = enrichment[description]["userListId"]
results = enrich(userListId, library, alpha)
enrichment[description]["libraries"][library] = results
break
except Exception as e:
print(e)
time.sleep(0.5)
else:
if description not in failed_enrich:
failed_enrich[description] = []
failed_enrich[description].append(library)
continue
time.sleep(0.1)
if len(failed_userlist):
print("Failed to add %d list"%len(failed_userlist))
if len(failed_enrich):
print("Failed enrichment for %d gene sets"%len(failed_enrich))
</code>
<code>
for lib in libraries:
display(Markdown("## %s"%lib.replace("_"," ")), display_id="title_%s"%lib)
term_df,table = get_dataframe(enrichment, lib, table, display_id=lib)
consensus, table = get_consensus(term_df, lib, top_results, table, display_id=lib)
# Visualize
consensus_df = term_df.loc[consensus.index]
if (consensus_df.shape[1] > 0):
clustergram_filename = "%s_consensus_clust.tsv"%lib
clustergram_caption = "Clustergrammer for the top %d consensus terms for %s "%(top_results, lib.replace("_"," "))
clustergrammer(consensus_df,
clustergram_filename,
clustergrammer_url,
lib,
figure,
clustergram_caption,
)
figure+=1
results_count = len(consensus.index) if len(consensus.index) < top_results else top_results
heatmap(consensus_df, "%s_consensus.svg"%lib, lib, width, height)
display(Markdown("**Figure %d** Heatmap for the top %d consensus terms for %s. [Download figure](%s_consensus.svg)"%(figure, results_count, lib.replace("_"," "), lib)),
display_id="heatmap_caption_%s"%lib)
figure+=1
# if num_sigs <=15:
status = stackedBarPlot(consensus_df, "%s_consensus_barplot.svg"%lib, display_id=lib)
if status:
display(Markdown("**Figure %d** Stacked bar plot for the top %d consensus terms for **%s**. [Download figure](%s_consensus_barplot.svg)"%(figure, top_results, lib.replace("_"," "), lib)),
display_id="stacked_bar_caption_%s"%lib)
figure +=1
else:
print("No terms found")
</code>
## References
[1] Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;128(14).
[2] Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016; gkw377.
[3] Fernandez, N. F. et al. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 4:170151 doi: 10.1038/sdata.2017.151 (2017).
|
{
"filename": "EnrichrConsensus_1.ipynb",
"repository": "MaayanLab/appyter-catalog",
"query": "transformed_from_existing",
"size": 23367,
"sha": ""
}
|
# main.ipynb
Repository: ZenVInnovations/9.-enhancing-text-analytics-data-quality-with-nlp---d24f3a13
<code>
!pip uninstall -y numpy thinc spacy torch
</code>
<code>
!pip install numpy==1.26.4
!pip install torch==2.2.2
!pip install spacy==3.7.2 thinc==8.2.2
!pip install nltk textblob
</code>
<code>
!python -m spacy download en_core_web_sm
</code>
<code>
import re
import spacy
from textblob import TextBlob
import nltk
from nltk.corpus import stopwords
# Download nltk stopwords if not already done
nltk.download('stopwords')
# Load spaCy model for NER
nlp = spacy.load('en_core_web_sm')
# Sample raw text (e.g. customer review or tweet)
raw_text = """
I LOVE this product!!! 😍😍 It's amazing. Bought it from Amazon last week.
Delivery was quick. Highly recommended. #happycustomer @amazon
"""
# 1. Text Normalization
def normalize_text(text):
# Lowercase
text = text.lower()
# Remove URLs
text = re.sub(r'http\S+|www\S+|https\S+', '', text, flags=re.MULTILINE)
# Remove mentions and hashtags
text = re.sub(r'@\w+|#\w+', '', text)
# Remove emojis and special characters (basic)
text = re.sub(r'[^\w\s]', '', text)
# Remove extra whitespace
text = re.sub(r'\s+', ' ', text).strip()
# Remove stopwords
stop_words = set(stopwords.words('english'))
filtered_words = [w for w in text.split() if w not in stop_words]
return ' '.join(filtered_words)
normalized_text = normalize_text(raw_text)
print("Normalized Text:", normalized_text)
# 2. Named Entity Recognition (NER)
doc = nlp(normalized_text)
entities = [(ent.text, ent.label_) for ent in doc.ents]
print("Named Entities:", entities)
# 3. Sentiment Analysis
sentiment = TextBlob(normalized_text).sentiment
print(f"Sentiment Polarity: {sentiment.polarity:.3f}, Subjectivity: {sentiment.subjectivity:.3f}")
# Validate sentiment
if sentiment.polarity > 0:
print("Overall Sentiment: Positive")
elif sentiment.polarity < 0:
print("Overall Sentiment: Negative")
else:
print("Overall Sentiment: Neutral")
</code>
<code>
import re
import spacy
from textblob import TextBlob
import nltk
from nltk.corpus import stopwords
# Download stopwords if not already downloaded
nltk.download('stopwords')
# Load spaCy model
nlp = spacy.load('en_core_web_sm')
# Text normalization function
def normalize_text(text):
text = text.lower()
text = re.sub(r'http\S+|www\S+|https\S+', '', text, flags=re.MULTILINE)
text = re.sub(r'@\w+|#\w+', '', text)
text = re.sub(r'[^\w\s]', '', text)
text = re.sub(r'\s+', ' ', text).strip()
stop_words = set(stopwords.words('english'))
filtered_words = [w for w in text.split() if w not in stop_words]
return ' '.join(filtered_words)
# Interactive loop
while True:
print("\n--- Text Analytics Interactive Tool ---")
user_input = input("Enter your text (or type 'exit' to quit):\n> ")
if user_input.lower() == 'exit':
break
normalized_text = normalize_text(user_input)
print("\nNormalized Text:", normalized_text)
doc = nlp(normalized_text)
entities = [(ent.text, ent.label_) for ent in doc.ents]
print("Named Entities:", entities if entities else "No named entities found.")
sentiment = TextBlob(normalized_text).sentiment
print(f"Sentiment Polarity: {sentiment.polarity:.3f}, Subjectivity: {sentiment.subjectivity:.3f}")
if sentiment.polarity > 0:
print("Overall Sentiment: Positive")
elif sentiment.polarity < 0:
print("Overall Sentiment: Negative")
else:
print("Overall Sentiment: Neutral")
</code>
|
{
"filename": "main.ipynb",
"repository": "ZenVInnovations/9.-enhancing-text-analytics-data-quality-with-nlp---d24f3a13",
"query": "transformed_from_existing",
"size": 35326,
"sha": ""
}
|
# Comparison_in_single-cell_data-checkpoint_3.ipynb
Repository: Velcon-Zheng/DL-mo
# SUB-BENCHMARK3: Comparing jDR methods on single-cell datasets
The performances of the 9 jDR methods are here compared based on their ability to cluster cells based on their cancer cell line of origine. The clustering is performed jointly considering scRNA-seq and scATAC-seq data.
## Data preprocessing
First the data are read in their original format and adapted to be read as input of our run_factorization function.
<code>
# Load data and processing
# Load RNA-seq data
exp <- readRDS("../data/single-cell/CellLines_RNAseqCounts.RDS", refhook = NULL) #ENS for genes and counts
# Apply log2 on RNA-seq data
exp <- log2(exp+1)
# Load ATAC-seq data
atac_counts<-readRDS("../data/single-cell/CellLines_ATACseqCounts.RDS", refhook = NULL) # peaks counts
# Load metadata
metadata<-readRDS("../data/single-cell/CellLines_metadata.RDS", refhook = NULL)
# Rename columns from metadata
colnames(atac_counts) <- metadata[,1]
# Export RNA-seq data as tab-separated table
write.table(exp, "../data/single-cell/CellLines_RNAseqCounts.txt",
sep="\t", col.names=TRUE, row.names=TRUE)
# Add a name ("probe") to the first column
system("sed -i '1s/^/probe\t/' ../data/single-cell/CellLines_RNAseqCounts.txt")
# Export ATAC-seq data as tab-separated table
write.table(atac_counts, "../data/single-cell/CellLines_ATACseqCounts.txt",
sep="\t", col.names=TRUE, row.names=TRUE)
# Add a name ("probe") to the first column
system("sed -i '1s/^/probe\t/' ../data/single-cell/CellLines_ATACseqCounts.txt")
</code>
## Running comparison
Two factor are then detected for each jDR method and the distribution of the cells with respect of Factor1 and Factor2 is plotted as a scatter plot. The obtained plots are available in the Results folder. The capability of the different jDR methods to cluster the cells accoridng to their cell line of origin is finally evaluated through the C-index, whose value is reported in the Results folder.
<code>
library("ggplot2")
library("clusterCrit")
source("runfactorization.R")
# Parameters for the plots
dot_size <- 1.5
dot_alpha <- 1.0
xlabel <- "Factor 1"
ylabel <- "Factor 2"
# Load annotations from the metadata
sample_annot <- metadata[, c("sample.rna", "celltype")]
# Folder for results
results_folder <- "../results_single_cell/"
# Create output folder
dir.create(results_folder, showWarnings = FALSE)
# Run factorization methods
out <- runfactorization("../data/single-cell/",
c("CellLines_RNAseqCounts.txt", "CellLines_ATACseqCounts.txt"),
2,
sep="\t",
filtering="stringent")
c_index <- numeric(0)
# For each factorization method
for(i in 1:length(out$factorizations)){
# Get factorization result
factors <- out$factorizations[[i]][[1]]
# Delete NAs
factors <- factors[!is.na(factors[,1]) & !is.na(factors[,2]), ]
sample_annot <- sample_annot[!is.na(sample_annot[,1]) & !is.na(sample_annot[,2]), ]
# Data to be plotted
df <- data.frame(x = factors[,1], y = factors[,2], color_by = sample_annot[,2])
# Plot results
p <- ggplot(df, aes_string(x = "x", y = "y")) +
geom_point(aes_string(color = "color_by"), size=dot_size, alpha=dot_alpha) +
xlab(xlabel) + ylab(ylabel) +
# scale_shape_manual(values=c(19,1,2:18)[seq_along(unique(shape_by))]) +
theme(plot.margin = margin(20, 20, 10, 10),
axis.text = element_text(size = rel(1), color = "black"),
axis.title = element_text(size = 16),
axis.title.y = element_text(size = rel(1.1), margin = margin(0, 10, 0, 0)),
axis.title.x = element_text(size = rel(1.1), margin = margin(10, 0, 0, 0)),
axis.line = element_line(color = "black", size = 0.5),
axis.ticks = element_line(color = "black", size = 0.5),
panel.border = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
legend.key = element_rect(fill = "white"),
legend.text = element_text(size = 16),
legend.title = element_text(size =16)
)
p + scale_color_manual(values=c("#0072B2", "#D55E00", "#CC79A7"))
# Export plot as JPEG image
ggsave(paste0(results_folder, "plot_",out$method[i],".jpg"))
# Encode cell type annotations by numeric codes
ann <- factor(sample_annot[,2], levels=c("HCT", "Hela", "K562"))
ann <- as.integer(ann)
# Compare factors and annotations
c_index <- c(c_index, intCriteria(factors, as.integer(ann), crit=c("C_index"))$c_index)
}
# Build output table
report_cindex <- data.frame(method=out$method, cindex=c_index)
# Export results as one tab-separated table
write.table(report_cindex, file = paste0(results_folder, "singlecell_cindex.txt"),
sep="\t", col.names=FALSE, row.names=FALSE, quote=FALSE)
</code>
|
{
"filename": "Comparison_in_single-cell_data-checkpoint_3.ipynb",
"repository": "Velcon-Zheng/DL-mo",
"query": "transformed_from_existing",
"size": 12497,
"sha": ""
}
|
# Report.ipynb
Repository: 27410/group-assingment-team11
# Biosynthesis of Isoamyl Acetate by Saccharomyces cerevisiae
## 1. Introduction
### 1.1 Literature review of the compound (<500 words)
### Overview on the product
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/iso_ace_pathway.jpg" alt="Pathway" width="350">
<figcaption>Figure 1. Pathway for the synthesis of isoamyl acetate</figcaption>
</figure>
Esters are amongst the most important flavour compounds in alcoholic beverages (Quilter et al., 2023). They are formed as byproducts in the reaction of esterification of alcohols with acetyl coenzyme A (see Figure 1) (Yoshimoto and Bogaki, 2023). One of the principal esters produced during alcohol fermentation is isoamyl acetate which gives a banana-like flavour (Furukawa, 2012). Isoamyl acetate is produced from a reaction between amyl alcohol and acetyl coenzyme A catalyzed by the enzyme isoamyl alcohol acetyl transferase (Yoshimoto and Bogaki, 2023).
#### Applications of the product
Isoamyl acetate is a flavoring and fragrance additive in various products because of its fruity banana flavor and aroma (Lopes et al., 2017). It is used as an additive to food like different candies which try to emulate the sweet taste of bananas (Mordor Intelligence, 01.12.2013). It is added to several alcoholic beverages as a way to add complexity to wines (Lopes et al., 2017). Even the pharmaceutical industry uses isoamyl acetate as an additive to make chewing tablets more attractive and easier for children to eat (Mordor Intelligence, 01.12.2013). Another application of isoamyl acetate include cosmetics and personal care industry as an odor additive to products like perfume and body creams (Torres, et al., 2010).
#### Market potential
From market observations done in the period between 2018 and 2023, the CAGR of isoamyl acetate was estimated to be above 3% (Mordor Intelligence, 01.12.2013). This means there's a growing and healthy global demand for this compound in multiple industries. The main growing market for this compound has been the Asian-Pacific market where personal care interest has skyrocketed in the last few years. The Asian-Pacific market's global share has been estimated to be 45% of all isoamyl acetate traded. The main countries with growing industries needing isoamyl acetate are China, Japan, India, and South Korea (Mordor Intelligence, 01.12.2013).
A future market can also be estimated to exist in countries like Vietnam. This is because of its growing population and cheap labor which is, for some, a more enticing alternative to China. This pivot of production and labor away from China is because of recent hostilities with China and the risks totalitarian regimes like China can have to business (Mai and Phuong, 2020).
### 1.2 Literature review of the cell factory (<500 words)
Fermentation, typically conducted under mild conditions, is energy-efficient and has a lesser environmental footprint than conventional chemical processes (Nielsen and Jegannathan, 2013). Therefore microbial biosynthesis of isoamyl acetate is preferred for its sustainability, utilizing cell factories capable of converting various substrates, such as agro-industrial by-products, into high-value products (Zhang et al., 2022).
Over millennia, yeast Saccharomyces cerevisiae (S. cerevisiae) has functioned as a cellular manufacturing hub for the synthesis of staple commodities such as bread, beer, and wine. In more recent times, S. cerevisiae has transcended its traditional applications, assuming the role of a versatile cell factory for the production of diverse fuels, chemicals, food constituents, and pharmaceuticals (Nielsen, 2019). Moreover, S. cerevisiae stands as a pivotal model organism in elucidating eukaryotic biology. Presently, myriad advancements crucial to the understanding and treatment of human diseases continue to emerge through investigations utilizing S. cerevisiae as a model organism (Nielsen, 2019).
Saccharomyces cerevisiae is our preferred host for isoamyl acetate biosynthesis, leveraging its natural ester-producing capabilities showcased in alcoholic fermentation processes. Genetic optimization of this yeast can improve precursor conversion efficiency, drawing from its established industrial utility and extensive research background (Singh et al., 2008).
## 2. Problem definition (<300 words)
Product-related issues include low production rates, which limit industrial scalability and economic feasibility (Yuan, 2016). Additionally, the concentrations of isoamyl acetate achieved in fermentation broths are often suboptimal, necessitating costly downstream processing to reach commercially viable levels (Mansor, 2020). Another concern is the low yield of isoamyl acetate relative to the glucose consumed, which reflects inefficiencies in the metabolic pathway of S. cerevisiae (Yuan, 2016).
Host cell-related problems present additional hindrances. Slow growth rates of S. cerevisiae can delay production timelines, impacting throughput. The toxicity of isoamyl acetate at higher concentrations can also impair cell health, further reducing productivity (Saerens, 2010).
This project aims to address these challenges through metabolic engineering strategies to enhance the growth rate and robustness of S. cerevisiae and increase the production rate, concentration, and yield of isoamyl acetate. By optimizing the metabolic pathways and fermentation processes, we intend to develop a more efficient, cost-effective, and scalable production method for high-quality isoamyl acetate.
## 3. *If Project category II:* Selection and assessment of existing GSM (<500 words)
Genome-scale metabolic models (GEMs) represent extensive knowledgebases that provide a platform for model simulations and integrative analysis of omics data. Our choice of host organism, S. cerevisiae, is a very common and well-researched organism, thus multiple genome-scale metabolic models (GSMs) exist. In order to choose the most optimal GSM available, 6 potential S. cerevisiae models were obtained form BioModels iFF708, iLL672, iMM904, iND750, yeast_7.6 and yeast8.
The first S. cerevisiae model to be established, iFF708, integrates 1175 reactions and 733 metabolites (Förster et al., 2003). It has been noted for accurate prediction of various cellular functions (Famili et al., 2003) and single gene deletion forecasts (Förster et al., 2003). The subsequent model, iND750, distinguishes itself with full compartmentalization and includes 750 genes and their associated transcripts, proteins, and reactions (Duarte et al., 2004). Its validation came from extensive gene deletion studies and analysis of metabolic phenotypes (Duarte et al., 2004), and it further incorporates regulatory elements to predict gene expression and outcomes in various transcription factor mutants (Herrgard et al., 2006). The third model, iLL672, is an evolution of iFF708; addresses and removes several dead-end reactions found in iFF708, thereby refining the accuracy of predictions for single gene deletions (Kuepfer et al., 2005). The iMM904 metabolic network was reconstructed based on iND750 to investigate how changes in the extracellular metabolome can be used to study systemic changes in intracellular metabolic states (Mo et al., 2009). It has shown consistency in the predicted intracellular flux changes with published measurements on intracellular metabolite levels and fluxes (Mo et al., 2009). Furthermore, model 7.6 incorporates more reactions, genes and metabolites than the previous model, iMM904 (Lu et al., 2019). The last, most up-to-date model developed was yeast8 (Lu et al., 2019).
Using memote, it is possible to assess different quality measures of the GSMs, including stochiometry, annotation, and reaction/metabolite statistics. The chosen models were compared using memote and the results are summarised in the table below.
| Measure | iFF708 | iND750 | iLL672 | iMM904 | yeast_7.6 | yeast8 |
| ---- | ---- | - | - | - | - | - |
| Total Metabolites | 796 | 1,061 | 671 | 1,228 | 3,370 | 2,806 |
| Total Reactions | 1,379 | 1,266 | 1,195 | 1,577 | 4,643 | 4,131 |
| Total Genes | 619 | 0 | 0 | 905 | 750 | 1,146 | 1,163 |
| Stochiometric Consistency | 0.0% | 100.0% | 0.0% | 100.0% | 0.0% | 0.0%|
| Mass Balance | 0.0%| 97.3%| 0.0% |0.0%|40.4%|93.8%|
| Metabolite Connectivity | 100.0% | 100.0% | 100.0% |100.0% | 100.0% | 100.0% |
| Metabolite Annotation | 0.0% | 0.0% | 0.0% | 0.0% | 100.0% | 100.0% |
| Reaction Annotation | 0.0% | 0.0% | 0.0% | 0.0% | 100.0% | 100.0% |
| Gene Annotation | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 100.0% |
| **Total score** |15% | 37% | 15% | 31% | 32% | 66% |
The report gave a structured overview on the most important aspects of the models. Yeast 8 and yeast 7.6 clearly overperformed the other models in terms of the number of metabolites, reactions, genes and annotations. Ultimately, yeast 8 was chosen due to it's highest total score and the presence of gene annotations, as opposed to yeast 7.6.
## 4. Computer-Aided Cell Factory Engineering (<1500 words for Category II project)
### 1. Theoretical yields and media optimization
The theoretical yield represents the amount of isoamyl acetate that can be obtained per amount of glucose added to the media. Theoretical productivites of biomass and isoamyl acetate for s. cerevisiae in the default medium are:
<!-- <center> -->
$0.03 \, \text{{biomass/h}} \, \text{{and}} \, 0.37 \, \text{{mmol isoamyl acetate/gDW}}\cdot\text{{h}}.$
<!-- </center> -->
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/Media_significance.png" alt="Media significance" width="900">
<figcaption>Figure 2. Productivities and yields of growth and isoamyl acetate
production when changing glucose concentration in the media</figcaption>
</figure>
In order to find the most optimal medium composition for our process, growth rate needs to be considered as well as the isoamyl acetate yield - as when having more cells more product is made overall.
Isoamyl acetate productivity and cell growth increases with higher glucose concentrations up till ~550mmol/L, it does not correspond to the changes in the yield - product per unit of glucose (see Figure 2). Noticeably, whereas isoamyl acetate yield stays relatively the same up till ~400mmol/L glucose, biomass yield starts decreasing already above ~100mmol\L glucose.
Additional simulation was run with leucine addition to the medium (at 100mmol/L glucose), which showed further increase in the product yield - from 0.35 to 0.36mmol isoamyl acetate/mmol glucose and growth - from 3.8\h to 7.62\h for 39mmol/L leucine . Addition of more leucine increases the product yield but compromises the growth.
The most optimal medium composition could be chosen by the substrate-cost-to-yield analysis and the actual fermentation experiments. However, this initial simulation gives a good overview on the possible glucose and leucine ranges to be tested.
See in depth analysis in Notebook [03_Theoretical_yields_and_media_optimization](03_Theoretical_yields_and_media_optimization.ipynb)
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/PPP.jpeg.jpg" alt="Phenotypa phase plane" width="900">
<figcaption>Figure 7. DFBA: Batch fermentation simulation results</figcaption>
</figure>
### 2. Phenotype phase plane
In the phenotype phase plane 4 different graphs were constructed, two focused on oxygens effect on biomass production and product formation, while the last two illustrated the glucose's effect on these. These simulations were done to find the optimum flux for biomass and Isoamyl acetate production.
It is logical to think that in order to achieve maximum biomass production and isoamyl acetate formation the cell must consume as much glucose as possible as fast as possible. However, when looking at the graphs we can see a different reality: when the flux of glucose increases above 579 mmol/gDW/h, the biomass production rate decreases substantially. Meanwhile, the isoamyl acetate formation rate begins to plateau at 450 and reaches its top point at 632 mmol/gDW/h, but it slowly decreases in efficiency afterward. Therefore, it can be concluded that the glucose uptake rate should be optimized for biomass since it has a minimal effect on product formation.
When evaluating the oxygen graphs, the same optimum flux can be observed on both graphs. This optimum is 316 mmol/gDW/h which then rapidly decreases afterwards. This rapid decrease in efficiency at high oxygen uptake rates may be due to oxidative stresses in the cell as a result of the high oxygen concentration inside the cell.
See in depth analysis in Notebook [04_Phenotype_phase_plane](04_Phenotype_phase_plane.ipynb)
### 3. Regulation gene targets
To optimize the cell factory for increased production of isoamyl acetate we performed FSEOF analysis, which stands for Flux Scanning based on Enforced Objective Flux, is a method used to identify which reactions within a metabolic network can have their fluxes adjusted to enhance the yield of a desired product.
#### Downregulation targets
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/Downregulation_targets.png" alt="Downregulated reactions" width="650">
<figcaption>Figure 4. FSEOF: Downregulated reactions</figcaption>
</figure>
Among downregulated reactions were highlighted following reactions will the lowest final flux in the range from -0.524184 to -0.500763, as more impactful.
1. Citrate transport (r_1126, final flux: -0.524184)
(S)-malate + citrate ==> (S)-malate + citrate
2. 2-isopropylmalate hydratase (r_0023, final flux: -0.504071)
2-isopropylmaleic acid + H2O ==> 2-isopropylmalate
3. 3-isopropylmalate dehydratase (r_0060, final flux: -0.504071)
(2R,3S)-3-isopropylmalate ==> 2-isopropylmaleic acid + H2O
4. 2-isopropylmalate transport (r_1574, final flux: -0.504071)
2-isopropylmalate ==> 2-isopropylmalate
5. Isoamyl alcohol transport (r_1864, final flux: -0.500763)
isoamylol ==> isoamylol
Downregulation of these reaction affects on enhanced utilization of recursors for isoamyl acetate production. Citrate transport and 2-isopropylmalate converstions optimize accumulation of acetyl-coA and leucin, respectively, those compounds are known as precursors for our product. It was, indeed, shown by Vadali et. al (2004) that increase in the intracellular levels of CoA and acetyl-Coenzyme A leads to higher isoamyl acetate production. Moreover, inhibiting isoamyl alchohol transport utilization for other reactions than isoamyl acetate production can result in target compound aggregation.
#### Upregulation targets
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/Overexpression_targets.png" alt=" Overexpression reactions" width="650">
<figcaption>Figure 5. FSEOF: Upregulated reactions</figcaption>
</figure>
Overexpression targets with the highest final flux in the range from 0.2068805 to 0.512811 can be theoretically adjusted to potentially boost the supply of essential precursors or energy needed for isoamyl acetate synthesis.
1. malic enzyme (r_0719, final flux: 0.512811)
(S)-malate + NADP(+) ==> carbon dioxide + NADPH + pyruvate
2. glycine transport (r_1811, final flux: 0.316483)
L-glycine ==> L-glycine
3. glycerol-3-phosphate dehydrogenase (r_0491, final flux: 0.206805)
dihydroxyacetone phosphate + H+ + NADH ==> glycerol 3-phosphate + NAD
Improving the functionality of malic enzyme and glycerol-3-phosphate dehydrogenase might significantly increase the availability of NADPH and NAD, respectively, which are vital co-factors necessary for the production of target compounds. Additionally, glycine is a key component in the synthesis of leucine, serving as the primary precursor in the leucine pathway.
<br>
<br>
---
However, choosing a target for downregulation or overexpression is not only about selecting the reaction with the highest flux change. It also depends on the metabolic pathway context, the connection of the reaction to isoamyl acetate production, and the potential metabolic burden that overexpression might cause. Additional experimental validation would be essential to confirm the benefits of downregulation or overexpressing this reaction in the context of the whole cell metabolism.
See in depth analysis in Notebook [05_Gene_target_analysis](05_Gene_target_analysis.ipynb)
### 4. Co-factor swap
The analysis of the isoamyl acetate synthesis pathway revealed NAD+-depended reaction of reducing isoamyl aldehyde (3-methylbutanal) to isoamyl alcohol (isoamylol) by aldehyde dehydrogenase:
r_0180: aldehyde dehydrogenase (isoamyl alcohol, NAD): s_0236(3-methyilbutanal) + s_0799 (glyoxylate)+ s_1205 (NADH) --> s_0931 (isoamylol) + s_1200 (NAD)
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/co-factor_swap_results.jpeg" alt="Co-factor Swap Results" width="500">
<figcaption>Figure 6. Co-factor swap results</figcaption>
</figure>
Isoamyl alcohol created in the above reaction is then transformed into isoamyl acetate in the reaction with Acetyl-CoA. Therefore, it was hypothesised that inceasing the availability of NAD+ can contribute to an increase in isoamyl acetate production. This would be achieved by changing NAD+/NADH utilization into NADP+/NADH in the chosen reactions in the metabolic pathways.
CofactorSwapOptimization by cameo was run on the model with its objective set to isoamyl acetate production. It reveled 4 NAD+/NADH-dependent reactions most suitable for co-factor swapping to benefit the model objective:
1. (S)-malate + NAD <=> H+ + NADH + oxaloacetate
2. dihydroxyacetone phosphate + H+ + NADH --> glycerol 3-phosphate + NAD
3. acetaldehyde + H+ + NADH --> ethanol + NAD
4. H+ + NADH + ubiquinone-6 --> NAD + ubiquinol-6
Co-factor swapping in the chosen reactions was executed by deleting the reaction genes and adding the reactions with the swapped co-factors to the model. Subsequently, the biomass production and isoamyl acetate productions were assessed. The analysis revealed ~70% and ~20% increase in biomass production and isoamyl acetate yield, respectively, as seen in FigX.
See in depth analysis in Notebook [06_Co-factor_swap](06_Co-factor_swap.ipynb)
<figure style="float: right; margin-left: 20px; text-align: center;">
<img src="Pictures/batch_fermentation_simulation.png" alt="Batch Fermentation Simulation" width="550">
<figcaption>Figure 7. DFBA: Batch fermentation simulation results</figcaption>
</figure>
### 5. Dynamic flux-based analysis
Dynamic flux-based analysis (DFBA) helps predict the growth and metabolic production rates over time, which is crucial for optimizing the conditions for cell growth and product formation.
Since phenotype phase plane analysis has shown the highest biomass and isoamyl acetate productivity with ~200 mmol/gDW*h, the fermentation was run in the aerobic conditions.
The simulation was visualized as seen in Figure 7. During the initial ~7.5h glucose declines sharply as it's consumed for cell growth and metabolism, dropping close to zero, coinciding with the drop of biomass production. The concentration of isoamyl acetate, on the other hand, increases steadily over time, even after the biomass reaches plateau. It makes sense, because we adjusted the lower bounds for the reaction and set the model objective to isoamyl acetate production. Ultimately, at the point of almost complete glucose depletion, the isaomyl acetate yield reaches 0.45mmol/L. It would require more simulations to uncover how the production would proceed upon the continuous addition of the substrate, liquid exchange, etc. or enriched medium. However, this initial DFBA gives a promising view on the fermentation in aerobic conditions with glucose as the sole carbon source.
See in depth analysis in Notebook [07_Dynamic_FBA](07_Dynamic_FBA.ipynb)
## 5. Discussion (<500 words)
The experimental process outlined in this study reveals potential solutions for boosting isoamyl acetate production in S. cerevisiae. So far, the most significant impact on yield has been achieved through simple medium recomposition; however, this solution may not be the most cost-effective in the long run. While higher glucose and leucine concentrations theoretically lead to increased biomass growth and product yield, practical constraints such as cost and potential inhibitory effects must be considered. Selecting optimal glucose and leucine concentrations becomes a decision influenced by both biological and economic factors.
On the other hand, cofactor swapping showed a ~1.2-fold increase in our compound production. Additionally, gene regulation targets suggest promising ways to increase substrate availability for isoamyl acetate production, with some proposed solutions related to existing literature (Vadali et al., 2004).
Phenotype phase plane analysis yielded interesting results. Biomass production was higher in the presence of oxygen, aligning with expectations. However, isoamyl acetate production also peaked at similar oxygen levels, contrary to literature data. Alcohol acetyltransferases, which catalyze reactions between alcohol and acetyl-CoA coenzyme, were shown to be repressed when yeasts are cultured under aerobic conditions (Torres, 2010). Thus, higher yields would be expected at lower oxygen levels. Nevertheless, DFBA was decided to be run in aerobic conditions, providing valuable predictive insights into the fermentation process.
The simulations in this project addressed isoamyl acetate yield and production rate, as well as host growth rates. The next steps for designing a functional cell factory would involve elucidating up- and downregulation targets and studying their influence on biomass and product yields. Moreover, fermentation experiments should be conducted with different media compositions, varying levels of glucose, leucine, and potentially pyruvate, all precursors in the isoamyl acetate pathway (Yoshimoto and Bogaki, 2023). Additionally, potential product accumulation toxicity should be considered, as it was not addressed in this report.
## 6. Conclusion (<200 words)
In conclusion, this study has highlighted several approaches to enhance isoamyl acetate production in S. cerevisiae. Medium recomposition emerges as a significant factor in yield improvement, yet its long-term cost-effectiveness remains a concern. Balancing the glucose and leucine concentrations presents a complex challenge, requiring consideration of both biological efficacy and economic viability. The promising results from co-factor swapping and targeted gene regulation, grounded in existing literature, offer viable paths forward. Intriguingly, our phenotype phase plane analysis contradicted some established literature by showing increased isoamyl acetate production under aerobic conditions, a finding that warrants further investigation.
The computational simulations provided valuable insights into the relationships between various factors influencing isoamyl acetate yield, production rate, and host growth rates.
## References
1. Duarte, N., Herrgård, M. & Palsson, B. Reconstruction and Validation of Saccharomyces cerevisiae iND750, a Fully Compartmentalized Genome-Scale Metabolic Model. *Genome research* **14**, 1298–309 (Aug. 2004).
2. Duarte, N., Palsson, B. & fu, P. Integrated analysis of metabolic phenotypes in Saccharomyces cerevisiae. *BMC genomics* **5**, 63 (Oct. 2004).
3. Famili, I., Forster, J., Nielsen, J. & Palsson, B. Saccharomyces cerevisiae phenotypes can be predicted by using constraint-based analysis of a genome-scale reconstructed metabolic network. *Proceedings of the National Academy of Sciences of the United States of America* **100**, 13134–9 (Nov. 2003).
4. Förster, J., Famili, I., Fu, P., Palsson, B. & Nielsen, J. Genome-Scale Reconstruction of the Saccharomyces cerevisiae Metabolic Network. *Genome research* **13**, 244–53 (Mar. 2003).
5. Förster, J., Famili, I., Palsson, B. & Nielsen, J. Large-Scale Evaluation of In Silico Gene Deletions in
Saccharomyces cerevisiae. *Omics : a journal of integrative biology* **7**, 193–202 (Feb. 2003).
6. Furukawa, S. *8 - Sake: quality characteristics, flavour chemistry and sensory analysis in Alcoholic Beverages*
(ed Piggott, J.) 180–195 (Woodhead Publishing, 2012). isbn: 978-0-85709-051-5.
7. Herrgård, M., Lee, B.-S., Portnoy, V. & Palsson, B. Integrated analysis of regulatory and metabolic networks
reveals novel regulatory mechanisms in Saccharomyces cerevisiae. *Genome research* **16**, 627–35 (June 2006).
8. Lu, H., Li, F., Sánchez, B.J. et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. *Nature Communications* **10**, 3586 (2019), doi:10.1038/s41467-019-11581-3.
9. Mordor *Acetate Market Size & Share Analysis - Growth Trends & Forecasts (2023 -2028)* Source: https://www.mordorintelligence.com/industry-reports/isoamyl-acetate-market https://www.
mordorintelligence.com/industry-reports/isoamyl-acetate-market. (Accessed: 01.12.2023).
10. Lopes, D. B., Madeira Júnior, J. V., de Castro Reis, L. V., Macena Leão, K. M. & Alves Macedo, G.
*Chapter 1 - Microbial Production of Added-Value Ingredients: State of the Art in Microbial Production of
Food Ingredients and Additives* (eds Holban, A. M. & Grumezescu, A. M.) 1–32 (Academic Press, 2017).
isbn: 978-0-12-811520-6.
11. Mai, N. & Phuong, P. Production Relocation of Multinational Companies from China and Chances for
Vietnam. *VNU JOURNAL OF ECONOMICS AND BUSINESS* **36** (2020).
12. Mansor, N., Singaravelan, R. & Abd Shukor, S. R. Parameters study on the production of isoamyl acetate
via milli-reactor in a solvent-free system. *IOP Conference Series: Materials Science and Engineering* **778**,
012067 (May 2020).
13. Mo, M., Palsson, B. & Herrgård, M. Connecting extracellular metabolomic measurements to intracellular
flux states in yeast. *BMC systems biology* **3**, 37 (Apr. 2009).
14. Nielsen J. (2019). Yeast Systems Biology: Model Organism and Cell Factory. *Biotechnology journal* **14**
15. Nielsen, P. & Jegannathan, K. R. Environmental assessment of enzyme use in industrial production: A
literature review. *Journal of Cleaner Production* (Nov. 2012).
16. Quilter, M., Hurley, J., Lynch, F. & Murphy, M. The Production of Isoamyl Acetate from Amyl Alcohol by
Saccharomyces cerevisiae. *Journal of the Institute of Brewing * **109** (Jan. 2003).
17. Saerens, S. M., Delvaux, F. R., Verstrepen, K. J., & Thevelein, J. M. (2010). Production and biological function of volatile esters in Saccharomyces cerevisiae. *Microbial biotechnology*, **3**(2), 165–177
18. Singh, R., Vadlani, P., Harrison, M., Bennett, G. & San, K.-Y. Aerobic production of isoamyl acetate by
overexpression of the yeast alcohol acetyl-transferases AFT1 and AFT2 in Escherichia coli and using low-cost fermentation ingredients. *Bioprocess and biosystems engineering* **31**, 299–306 (July 2008).
19. Torres, S., Pandey, A. & Castro, G. Banana flavor: Insights into isoamyl acetate production 225–244
(Jan. 2010). isbn: 978-1-61761-124-7.
20. Vadali, R., Bennett, G. & San, K.-Y. Enhanced Isoamyl Acetate Production upon Manipulation of the
Acetyl-CoA Node in Escherichia coli. *Biotechnology progress* **20**, 692–7 (June 2004).
<!--
18. Varman, A. M., Xiao, Y., Leonard, E. & Tang, Y. Statistics-based model for prediction of chemical biosynthesis yield from Saccharomyces cerevisiae. *Microbial cell factories* **10**, 45 (June 2011). -->
21. Yoshimoto, H. & Bogaki, T. Mechanisms of production and control of acetate esters in yeasts. *Journal of
Bioscience and Bioengineering* **136**, 261–269 (2023).
<!-- 23. Yoshioka, K. & Hashimoto, N. Ester Formation by Alcohol Acetyltransferase from Brewers’ Yeast. *Agricultural and Biological Chemistry* **45**, 2183–2190 (1981). -->
22. Yuan, J., Mishra, P. & Ching, C.-B. Engineering the leucine biosynthetic pathway for isoamyl alcohol
overproduction in Saccharomyces cerevisiae. *Journal of Industrial Microbiology & Biotechnology* **44** (Oct.
2016).
23. Zhang, Q., Xiao, N., Xu, H., Tian, Z., Li, B., Qiu, W. & Shi, W. Changes of Physicochemical Characteristics and Flavor during Suanyu Fermentation with Lactiplantibacillus plantarum and Saccharomyces cerevisiae. *Foods* **11**, 4085 (Dec. 2022).
|
{
"filename": "Report.ipynb",
"repository": "27410/group-assingment-team11",
"query": "transformed_from_existing",
"size": 34472,
"sha": ""
}
|
# CopyNumberEstimation-checkpoint.ipynb
Repository: dpeerlab/MitoEJ-paper-analysis
# Copy Number Estimation
This notebook estimates mtDNA copy number using the method developed at SAIL. It is based on the assumption that scATAC-seq samples open nuclear DNA at the same rate as mitchondrial DNA, which is open everywhere. We choose open bins in nuclear DNA by computing an accessibiity score for each bin, and choosing bins with high accessibility score. Then we use these bins as normalization factor to compute the mtDNA copy number.
<code>
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import argparse
import time
import scanpy as sc
import fastremap
from scipy.sparse import csr_matrix
import anndata
import bisect
from tqdm import tqdm
import os
plt.rcParams["figure.figsize"] = (15, 7)
</code>
# Step 1: Load data
<code>
def read_fragment_file(fragment_path):
"""
Read fragment file from the specified path.
"""
fragment_data = pd.read_csv(
fragment_path,
compression='gzip',
header=None,
sep='\t',
quotechar='"',
comment='#',
dtype={'chromosome': str} # Ensure 'chromosome' is read as string
)
fragment_data.columns = ['chromosome', 'start', 'end', 'barcode', 'cr_umi']
return fragment_data
</code>
<code>
def read_chromosome_size_file(reference):
"""
read chromosome size file for given reference
"""
return pd.read_csv(f'/data/peer/rsaha/mito/copy-number-pipeline/genome_files/{reference}/{reference}.csv')
</code>
<code>
def read_blacklist(reference):
"""
read chromosome size file for given reference
"""
return pd.read_csv(f'/data/peer/rsaha/mito/copy-number-pipeline/genome_files/{reference}/blacklist.csv')
</code>
<code>
samples = [
"YF-2721_48mix_10dGal_3d_mito_hLLLdogma_multiome",
"YF-2721_48mix_25dGlu_3d_mito_hLLLdogma_multiome",
"YF-2721_48mix_25dGlu_NoPyr_3d_mito_hLLLdogma_multiome",
]
sample_to_short_dict = {
"YF-2721_48mix_10dGal_3d_mito_hLLLdogma_multiome": 'Galactose',
"YF-2721_48mix_25dGlu_3d_mito_hLLLdogma_multiome": 'Glucose',
"YF-2721_48mix_25dGlu_NoPyr_3d_mito_hLLLdogma_multiome": 'Glucose_No_Pyruvate',
}
s = samples[2]
</code>
<code>
proj_dir = "/data/peer/landm/Projects/sail-dogma/yi-clone-samples/"
INBASE = f'{proj_dir}data/{s}/cr-arc-results/'
FRAGMENT_PATH = f'{INBASE}atac_fragments.tsv.gz'
BARCODE_PATH = f'{INBASE}filtered_feature_bc_matrix.h5'
OUTPUT_DIR = f'{proj_dir}notebooks/copy-number-estimation-cache/{sample_to_short_dict[s]}/'
GENOME = 'hg38'
</code>
<code>
if not os.path.exists(OUTPUT_DIR):
os.mkdir(OUTPUT_DIR)
</code>
<code>
frag = read_fragment_file(FRAGMENT_PATH)
frag.head()
</code>
<code>
barcodes = sc.read_10x_h5(BARCODE_PATH)
barcodes = pd.DataFrame(barcodes.obs.index, columns=['barcode'])
barcodes
</code>
<code>
chromosome_size = read_chromosome_size_file(GENOME)
chromosome_size
</code>
<code>
# filter fragments
frag = frag[frag['barcode'].isin(barcodes['barcode'])]
frag = frag[frag['chromosome'].isin(chromosome_size['Chromosome'])]
</code>
# Step 2: Compute Tile Matrix
## Read Blacklist Information
<code>
blacklist = read_blacklist(GENOME)
blacklist = blacklist[blacklist['chromosome'].isin(chromosome_size['Chromosome'])]
blacklist.head()
</code>
## Construct Tile Size
<code>
TILE_SIZE=500
</code>
<code>
def convert_to_linear_genome(frag):
offset = dict(zip(chromosome_size['Chromosome'], (np.ceil(np.array([0] + list(chromosome_size['length'][:-1]))//TILE_SIZE)*TILE_SIZE).astype(int).cumsum()))
chromosome_offset_list = np.array(list(map(lambda x: offset[x], frag['chromosome'])))
frag['start_linear'] = frag['start'] + chromosome_offset_list + 1
frag['end_linear'] = frag['end'] + chromosome_offset_list
</code>
## Convert blacklist to linear genome
<code>
convert_to_linear_genome(blacklist)
blacklist['start_bin'] = blacklist['start_linear']//TILE_SIZE
blacklist['end_bin'] = blacklist['end_linear']//TILE_SIZE
print('converted blacklist to linear genome')
</code>
## Convert fragment to linear genome
<code>
s = time.time()
convert_to_linear_genome(frag)
frag['start_bin'] = frag['start_linear']//TILE_SIZE
frag['end_bin'] = frag['end_linear']//TILE_SIZE
e = time.time()
print('converted fragment to linear genome', e - s , 'seconds')
</code>
## Remove blacklisted entries
<code>
def return_numpy_mask_remove_numbers_in_intervals(numbers, intervals):
# Step 1: Sort intervals based on the start of each interval
intervals.sort(key=lambda x: x[0])
# Step 2: Sort the list of numbers
# numbers.sort(key=lambda x: x[1])
# Step 3: Iterate using two pointers
interval_idx = 0
result = []
for idx, number in enumerate(numbers):
while interval_idx < len(intervals) and number[1] > intervals[interval_idx][1]:
interval_idx += 1
if interval_idx == len(intervals) or number[1] < intervals[interval_idx][0]:
result.append(idx)
# Step 4: Return the filtered list
return result
</code>
<code>
s = time.time()
intervals = list(zip(blacklist['start_bin'], blacklist['end_bin']))
f = pd.concat([frag['barcode'], frag['barcode']]).to_numpy()
b = pd.concat([frag['start_bin'], frag['end_bin']]).to_numpy()
numbers = np.column_stack((f, b))
numbers = numbers[np.argsort(numbers[:,1].astype(int))]
mask = return_numpy_mask_remove_numbers_in_intervals(numbers, intervals)
filtered = numbers[mask]
e = time.time()
print('removed blacklisted entries', e - s, 'seconds')
</code>
## Count reads per bin
<code>
barcode_to_index = pd.DataFrame(range(len(barcodes['barcode'])), index=list(barcodes['barcode']))
</code>
<code>
barcode_to_index.head()
</code>
<code>
filtered_barcode_to_index = barcode_to_index.loc[filtered[:,0]]
</code>
<code>
filtered_barcode_to_index
</code>
<code>
unique_filtered = (filtered_barcode_to_index[0] * (filtered[:,1].max() + 1) + filtered[:,1]).to_numpy().astype(int)
</code>
<code>
unique_filtered
</code>
<code>
s = time.time()
uniq, cts = fastremap.unique(unique_filtered, return_counts=True) # may be much faster than np.unique
e = time.time()
print(f'fastremap took {e-s} seconds')
</code>
<code>
m, n = ((uniq // (filtered[:,1].max() + 1)), uniq - (uniq // (filtered[:,1].max() + 1)) * (filtered[:,1].max() + 1))
</code>
## Construct AnnData
<code>
chromosome_size['num_bins'] = np.ceil(chromosome_size['length']/TILE_SIZE).astype(int)
linear_genome_length_in_bins = int(chromosome_size['num_bins'].sum())
</code>
<code>
mat = csr_matrix((cts, (m, n)), shape=(len(barcodes['barcode']), linear_genome_length_in_bins))
</code>
<code>
tile_mat_adata = anndata.AnnData(mat, obs=barcodes)
</code>
<code>
tile_mat_adata
</code>
<code>
chromosome_size['num_bins'].cumsum()
</code>
<code>
def get_tile_name_df():
tile_name_df = pd.DataFrame(list(zip(np.arange(0, tile_mat_adata.shape[1]) * TILE_SIZE, np.arange(0, tile_mat_adata.shape[1]) * TILE_SIZE + TILE_SIZE - 1)), columns=['start', 'end'])
tile_name_df['chromosome'] = np.nan
for i in range(len(chromosome_size)):
chromosome_name = chromosome_size.iloc[i]['Chromosome']
if i == 0:
bin_range_start = 0
else:
bin_range_start = chromosome_size['num_bins'].cumsum().iloc[i-1]
bin_range_end = chromosome_size['num_bins'].cumsum().iloc[i]
tile_name_df['chromosome'].iloc[bin_range_start: bin_range_end] = chromosome_name
return tile_name_df
</code>
<code>
tile_name_df = get_tile_name_df()
</code>
<code>
tile_name_df
</code>
<code>
tile_mat_adata.var = tile_name_df
</code>
<code>
tile_mat_adata
</code>
<code>
tile_mat_adata.write_h5ad(f'{OUTPUT_DIR}tile_matrix.h5ad')
</code>
# Step 3: Bin selection
<code>
def select_bins(pctl):
tile_accessibility_score = np.asarray(tile_mat_adata.X.sum(axis=0))[0]
threshold = np.quantile(np.array(tile_accessibility_score), pctl)
bins = np.array(tile_mat_adata[:,tile_accessibility_score > threshold].var.copy().index)
return bins
def subset_frag(bins):
bins = np.unique(np.concatenate([bins - 1, bins, bins + 1]))
frag_subset = frag[frag['start_bin'].isin(bins) | frag['end_bin'].isin(bins)]
return frag_subset
</code>
<code>
chromosome_size['num_bins'].cumsum()
</code>
<code>
bins = select_bins(0.985)
frag_subset = subset_frag(bins)
</code>
# Step 4: Construct Cell x Chromosome coverage matrix
<code>
frag_subset['length'] = frag_subset['end'] - frag_subset['start'] + 1
</code>
<code>
# remove frags in deleted region
mito_frags = frag_subset[frag_subset['chromosome'] == 'chrM']
</code>
<code>
del_start = 8013
del_end = 11460
remove_idx = mito_frags[(mito_frags['start'] > del_start) & (mito_frags['end'] < del_end)].index
</code>
<code>
frag_subset_filtered = frag_subset.drop(remove_idx)
</code>
<code>
print(frag_subset.shape[0])
print(frag_subset_filtered.shape[0])
</code>
<code>
def compute_cell_x_chromosome_coverage_df(frag):
pivot_df = pd.pivot_table(frag, values='length', index='barcode', columns='chromosome', aggfunc='sum', fill_value=0)
return pivot_df
</code>
<code>
cell_x_chromosome_coverage_df = compute_cell_x_chromosome_coverage_df(frag_subset_filtered)
</code>
<code>
cell_x_chromosome_coverage_df
</code>
<code>
cell_x_chromosome_coverage_df.to_csv(f'{OUTPUT_DIR}cell_x_chromosome_coverage_df.csv')
</code>
# Step 5: Compute copy number
<code>
mito_non_deleted_len = 16569 - (11460 - 8013)
</code>
<code>
def get_copy_estimates(cell_x_chromosome_coverage_df):
mito_coverage = cell_x_chromosome_coverage_df.values[:, -2]
nuclear_coverage = cell_x_chromosome_coverage_df.drop(columns=['chrM']).values
# mito_normalized_coverage = mito_coverage/chromosome_size[chromosome_size['Chromosome'] == 'chrM']['length'].iloc[0]
mito_normalized_coverage = mito_coverage/mito_non_deleted_len
nuclear_normalized_coverage = nuclear_coverage.sum(axis=1) / (len(bins) * TILE_SIZE)
copy_estimates = pd.DataFrame(2 / nuclear_normalized_coverage * mito_normalized_coverage, index = cell_x_chromosome_coverage_df.index, columns=['copy_number'])
return copy_estimates
</code>
<code>
copy_estimates = get_copy_estimates(cell_x_chromosome_coverage_df)
</code>
<code>
copy_estimates.to_csv(f'{OUTPUT_DIR}copy_estimates.csv')
</code>
<code>
copy_estimates['copy_number'].hist(bins=50, edgecolor='black')
</code>
|
{
"filename": "CopyNumberEstimation-checkpoint.ipynb",
"repository": "dpeerlab/MitoEJ-paper-analysis",
"query": "transformed_from_existing",
"size": 97739,
"sha": ""
}
|
# Adder.ipynb
Repository: BDR-Pro/QuiziWiki
<code>
# Connect to MongoDB
from pymongo.mongo_client import MongoClient
from pymongo.server_api import ServerApi
from dotenv import load_dotenv
import wikipedia as wp
import os
# Load environment variables from .env file
load_dotenv()
# Access an environment variable
password = os.getenv('MONGO')
uri = f"mongodb+srv://baderalotaibi3:{password}@cluster0.od393y9.mongodb.net/?retryWrites=true&w=majority"
# Create a new client and connect to the server
client = MongoClient(uri, server_api=ServerApi('1'))
db = client["WIKIQUIZ"]
collection = db["WikiQuizEnApi"]
PagesCollection = db["WikiPagesTitle"]
# Send a ping to confirm a successful connection
literature_topics = [
"The Evolution of the Novel",
"Shakespeare and the Elizabethan Era",
"The Gothic Novel",
"Romanticism in Literature",
"Victorian Literature",
"The Modernist Movement",
"Postmodern Literature",
"The Harlem Renaissance",
"Magical Realism and Its Pioneers",
"Dystopian Novels",
"The Beat Generation",
"The Golden Age of Detective Fiction",
"Science Fiction and Its Evolution",
"Fantasy Literature and World Building",
"The Bildungsroman Genre",
"Epic Poetry and Its Examples",
"The Influence of Greek Mythology on Literature",
"Feminism in Literature",
"Transcendentalism and American Literature",
"The Southern Gothic Tradition",
"The Bloomsbury Group",
"The Russian Greats: Dostoevsky, Tolstoy, and Others",
"Latin American Boom",
"African Literature and Postcolonialism",
"The Brontë Sisters",
"James Joyce and the Stream of Consciousness Technique",
"Existentialism in Literature",
"The Legacy of the French Nouveau Roman",
"The Rise of Autobiographical and Memoir Literature",
"Children's Literature and Its Evolution",
"The Role of the Antihero in Literature",
"Cyberpunk and Speculative Fiction",
"The Influence of Literature on Social Change",
"The Nobel Prize in Literature",
"The Booker Prize and Its Controversies",
"The Role of the Editor in Literature",
"The History of the Publishing Industry",
"Literary Criticism and Theory",
"Digital Literature and the Future of Reading",
"The Preservation of Endangered Languages through Literature",
"The Impact of Translation on World Literature",
"Oral Traditions and Their Influence on Written Literature",
"The Epistolary Novel",
"Satire and Political Commentary in Literature",
"Eco-Criticism and Nature Writing",
"The Bildungsroman in Contemporary Literature",
"Postcolonial Literature and Identity",
"Memoir vs. Autobiography",
"The Role of Libraries in Preserving Literature",
"Banned Books and Censorship",
"The Psychological Novel",
"The Renaissance in Literature",
"Neo-Classicism and Literature",
"The Romantic Hero in Literature",
"The Victorian Novel and Society",
"The Influence of War on 20th-Century Literature",
"Literature of the American Frontier",
"The Lost Generation Writers",
"The New Journalism Movement",
"Literary Journals and Their Impact",
"Flash Fiction and the Short Story",
"The Future of Print Books in a Digital Age",
"Fan Fiction and Literary Expansion",
"The Graphic Novel",
"Poetry in the Digital Age",
"The Rise of Young Adult Fiction",
"Indigenous Narratives and Storytelling",
"The Gothic Revival in Contemporary Literature",
"The Mystery Genre: From Poe to Present",
"Literary Festivals Around the World",
"The Impact of Self-Publishing",
"Literature and Mental Health",
"The Epistolary Novel in the 21st Century",
"Cross-Genre Literature",
"The Role of Myth in Literature",
"The Tragicomedy Genre",
"Literature as Resistance",
"The Influence of the Supernatural in Literature",
"The Development of the Essay",
"The History and Impact of Literary Salons",
"Literary Prizes and Their Influence on the Canon",
"The Evolution of Narrative Techniques",
"Epic Novels and Their Structure",
"The Role of Ghostwriters in Literature",
"The Significance of Cover Art in Literature",
"The Decadent Movement",
"Literary Hoaxes and Controversies",
"The Importance of Setting in Literature",
"The Concept of the Literary Canon",
"The Intersection of Literature and Philosophy",
"Autofiction and Its Rise",
"Literature and Environmental Awareness",
"The Influence of Technology on Writing and Reading",
"Narrative Nonfiction",
"Literature and Globalization",
"The Sociology of Literature",
"Literary Tourism and Pilgrimages",
"The Role of Critique Groups and Workshops in Writing",
"Serial Novels and Their Revival",
"The Impact of Literary Awards",
"The Evolution of the Biography",
"Fan Fiction: Legal and Literary Implications",
"Digital Archives and the Accessibility of Classic Texts",
"The Role of Book Clubs in Literary Culture",
"Interactive Fiction and Game Narratives",
"The Influence of Social Media on Literature",
"The Phenomenon of Literary Podcasts",
"Crowdsourced Writing and Literature",
"The Role of Literary Agents in the Modern Publishing Landscape",
"Literature in Education: Approaches and Controversies",
"Adaptations: From Books to Film and Beyond",
"The Economics of Book Publishing",
"Literary Scandals and Their Impact on Public Perception",
"The Future of Literary Criticism",
"E-literature and Its Platforms",
"The Intersection of Art and Literature",
"The Revival of Poetry Slams and Spoken Word",
"The Role of Zines in Underground Literature",
"The Influence of Celebrity Book Clubs",
"The Preservation of Literary Heritage",
"Emerging Voices in Contemporary Literature",
"The Global Market for Translated Literature",
"Literature and the Exploration of Gender",
"The Changing Landscape of Book Retail",
]
list_ = [
"Earth",
"Artificial intelligence",
"Astronomy",
"Science",
"Quantum Computing",
"Evolutionary Biology",
"Cybersecurity",
"Renewable Energy",
"Ancient Civilizations",
"Deep-sea exploration",
"Augmented reality",
"Machine Ethics",
"Space Colonization",
"Neuroscience",
"Consciousness",
"Chess",
"The History of the Internet",
"Black Holes and Their Mysteries",
"The Evolution of Video Games",
"Artificial Intelligence and Machine Learning",
"The Cultural Impact of The Beatles",
"Quantum Computing",
"The Lost City of Atlantis",
"The Life and Theories of Albert Einstein",
"The French Revolution",
"The Human Genome Project",
"Ancient Egyptian Gods and Goddesses",
"The Mystery of the Bermuda Triangle",
"The Art and Science of Cryptography",
"Climate Change and Its Global Impact",
"The Rise and Fall of the Roman Empire",
"The Deep Web and Dark Web",
"Space Exploration and the Mars Missions",
"The World of Dreams and Their Interpretations",
"Nikola Tesla and His Inventions",
"The Samurai of Japan",
"The Chernobyl Disaster",
"The Theory of Relativity",
"The Psychology of Happiness",
"The Great Wall of China",
"Leonardo da Vinci's Inventions",
"The Salem Witch Trials",
"The Life Cycle of Stars",
"The History of Chocolate",
"The Legend of King Arthur",
"The Development of the English Language",
"The Voyager Golden Record",
"The Fibonacci Sequence in Nature",
"The Cultural Significance of Tattoos",
"The Discovery of Penicillin",
"The Concept of Time Travel",
"The Rosetta Stone and Deciphering Ancient Scripts",
"The Impact of Social Media on Society",
"The Titanic: Before and After the Iceberg",
"Quantum Mechanics and its Paradoxes",
"The Mystery of Stonehenge",
"The Process of Natural Selection",
"The Apollo Moon Landings",
"The Psychology of Fear",
"The History of Pirates and Privateers",
"The Phenomenon of Sleep Paralysis",
"The Architectural Wonders of the Ancient World",
"The Origin and Spread of Coffee Culture",
"The Lives of the Pharaohs of Egypt",
"The Creation and Significance of the Silk Road",
"The Mysteries of the Human Brain",
"The History of the Olympic Games",
"The Great Depression",
"The Cultural Practices of the Maori of New Zealand",
"The Concept and Challenges of Sustainable Living",
"The Lore of Norse Mythology",
"The Dynamics of Black Holes",
"The Artistic Genius of Vincent van Gogh",
"The Evolution of the Computer",
"The Mystery of the Loch Ness Monster",
"The Science of Genetics and DNA",
"The History of the Samurai",
"The Story of Anne Frank",
"The Exploration of the Amazon Rainforest",
"The Development of the Atomic Bomb",
"The Impact of the Industrial Revolution",
"The Phenomenon of Bioluminescence in Nature",
"The Cultural Revolution in China",
"The Secrets of the Dead Sea Scrolls",
"The World of Quantum Physics",
"The Legend of the Holy Grail",
"The Impact of Globalization",
"The Mysteries of Antarctica",
"The Rise of Cryptocurrencies",
"The Architectural Marvels of Gaudi",
"The Phenomenon of Aurora Borealis (Northern Lights)",
"The History and Art of Origami",
"The Psychology Behind Dreams",
"The Wonders of the Galápagos Islands",
"The Story Behind the Invention of the Internet",
"The Traditions and History of Tea in China",
"The Mystery of Easter Island",
"The Science Behind Earthquakes and Volcanoes",
"The Tales of Greek Mythology",
"The History of the United Nations",
"The Concept of Virtual Reality",
"The Art of War by Sun Tzu",
"The Discovery of Pluto and Its Demotion",
"The Cultural Impact of Manga and Anime",
"The Science of Climate Change",
"The History of Chess",
"The Biology of Aging",
"The Process of Wine Making",
"The History of Espionage",
"The Development and Impact of the Printing Press",
"The Exploration of the Deep Ocean",
"The Innovations of the Renaissance Period",
"The History of Mathematics",
"The Secrets of the Mayan Civilization",
"The Theory of Everything in Physics",
"The Impact of Artificial Intelligence on Future Jobs",
"Virtual Reality and Its Applications",
"The Future of Transportation (Hyperloop, Electric Vehicles)",
"Smart Cities and Urban Planning",
"The Human Microbiome and Its Impact on Health",
"Advances in 3D Printing Technologies",
"The Psychology of Social Networks",
"Blockchain Technology and Its Uses Beyond Cryptocurrency",
"The Impact of Climate Change on Biodiversity",
"The Exploration of Mars: Rovers, Landers, and Future Missions",
"The Art and Science of Meditation",
"The Rise of eSports and Competitive Gaming",
"Genetic Engineering and CRISPR",
"The History and Future of the Internet of Things (IoT)",
"Sustainable Agriculture and Food Security",
"The Study of Exoplanets and the Search for Extraterrestrial Life",
"The Role of Nanotechnology in Medicine",
"The History of Artificial Intelligence",
"Renewable Energy Sources and Their Development",
"The Importance of Bees in Ecosystems",
"The Psychology of Learning and Memory",
]
science_topics = [
"Quantum Mechanics",
"General Relativity",
"The Big Bang Theory",
"Photosynthesis",
"DNA Replication",
"Black Holes",
"The Periodic Table",
"Evolution by Natural Selection",
"Plate Tectonics",
"The Higgs Boson",
"Vaccination and Immunology",
"The Theory of Evolution",
"Climate Change",
"Renewable Energy",
"Artificial Intelligence",
"Machine Learning",
"Conservation of Energy",
"Thermodynamics",
"The Scientific Method",
"Molecular Biology",
"Neuroscience",
"Psychology",
"Cognitive Science",
"Quantum Computing",
"Nanotechnology",
"The Human Genome Project",
"Stem Cell Research",
"Biodiversity",
"Ecology",
"Global Warming",
"Astronomy",
"Astrophysics",
"Cosmology",
"Dark Matter",
"Dark Energy",
"Exoplanets",
"The Solar System",
"The Milky Way",
"Galaxies",
"The Universe",
"Particle Physics",
"Atomic Structure",
"Chemical Reactions",
"Organic Chemistry",
"Inorganic Chemistry",
"Biochemistry",
"Physical Chemistry",
"Analytical Chemistry",
"The Cell",
"Genetics",
"Evolutionary Psychology",
"Botany",
"Zoology",
"Microbiology",
"Paleontology",
"Geology",
"Meteorology",
"Oceanography",
"Environmental Science",
"Quantum Field Theory",
"String Theory",
"Nuclear Fusion",
"Nuclear Fission",
"Renewable Resources",
"Non-renewable Resources",
"Sustainable Development",
"Human Anatomy",
"Physiology",
"Pathogens and Disease",
"Virology",
"Bacteriology",
"Mycology",
"Parasitology",
"Pharmacology",
"Toxicology",
"Endocrinology",
"Genomics",
"Proteomics",
"Metabolomics",
"Biotechnology",
"Genetic Engineering",
"CRISPR",
"Robotics",
"Cybersecurity",
"Data Science",
"Cryptography",
"Quantum Cryptography",
"The Internet",
"Blockchain Technology",
"Virtual Reality",
"Augmented Reality",
"Computer Vision",
"Natural Language Processing",
"Speech Recognition",
"Computational Biology",
"Mathematical Models in Science",
"Statistical Methods in Research",
"Optics",
"Acoustics",
"Fluid Dynamics",
"Solid State Physics",
"Electromagnetism",
"Classical Mechanics",
"Quantum Mechanics Applications",
"Materials Science",
"Polymers",
"Ceramics",
"Composites",
"Metallurgy",
"Quantum Chemistry",
"Structural Biology",
"Systems Biology",
"Evolutionary Biology",
"Synthetic Biology",
"Conservation Biology",
"Ecological Modelling",
"Hydrology",
"Atmospheric Sciences",
"Planetary Science",
"Geochemistry",
"Geophysics",
"Seismology",
"Volcanology",
"Paleoclimatology",
"Glaciology",
"Limnology",
"Soil Science",
"Agricultural Science",
"Forestry",
"Horticulture",
"Entomology",
"Ichthyology",
"Herpetology",
"Ornithology",
"Mammalogy",
"Primatology",
"Conservation Efforts",
"Wildlife Management",
"Marine Biology",
"Coral Reef Ecology",
"Deep Sea Biology",
"Fisheries Science",
"Aquaculture",
"Phylogenetics",
"Population Genetics",
"Molecular Evolution",
"Behavioral Ecology",
"Community Ecology",
"Ecosystem Services",
"Biogeochemical Cycles",
"Human Impact on the Environment",
"Climate Modelling",
"Renewable Energy Technologies",
"Solar Power",
"Wind Energy",
"Hydropower",
"Geothermal Energy",
"Biofuels",
"Nuclear Power",
"Energy Storage Technologies",
"Carbon Capture and Storage",
"Sustainable Cities",
"Green Chemistry",
"Environmental Policy",
"Waste Management",
"Recycling Technologies",
"Water Purification",
"Air Pollution Control",
"Land Reclamation",
"Contaminated Site Remediation",
"Invasive Species Control",
"Endangered Species Recovery",
"Habitat Restoration",
"Climate Change Mitigation",
"Climate Change Adaptation",
"Sustainable Agriculture",
"Precision Agriculture",
"Organic Farming",
"Urban Farming",
"Genetically Modified Organisms (GMOs)",
"Pesticides and Herbicides",
"Soil Conservation",
"Water Resources Management",
"Irrigation Technologies",
"Aquaponics and Hydroponics",
"Food Security",
"Nutritional Science",
"Diet and Health",
"Food Processing and Preservation",
"Fermentation Science",
"Brewing Science",
"Sensory Analysis of Food",
"Food Safety and Regulations",
"Public Health and Epidemiology",
"Vaccine Development",
"Antibiotic Resistance",
"Emerging Infectious Diseases",
"Global Health Initiatives",
"Healthcare Technologies",
"Biomedical Engineering",
"Medical Imaging",
"Regenerative Medicine",
"Precision Medicine",
"Telemedicine",
"Psychiatry and Mental Health",
"Neurodegenerative Diseases",
"Cancer Research",
"Cardiovascular Diseases",
"Diabetes Mellitus",
"Respiratory Diseases",
"Infectious Disease Therapies",
"Surgical Techniques",
"Anesthesia and Pain Management",
"Orthopedics",
"Dentistry and Oral Health",
"Pediatrics",
"Geriatrics",
"Nursing and Patient Care",
"Healthcare Policy and Management",
"Medical Ethics",
"Biostatistics",
"Clinical Trials",
"Epidemiological Studies",
"Health Informatics",
"Molecular Diagnostics",
"Pharmaceutical Sciences",
"Drug Discovery and Development",
"Pharmacokinetics and Pharmacodynamics",
"Medicinal Chemistry",
"Pharmacy Practice",
"Alternative Medicine",
"Physical Therapy] and Rehabilitation",
"Occupational Health and Safety",
"Environmental Health",
"Radiation Biology",
"Toxicological Sciences",
"Veterinary Sciences",
"Animal Behavior",
"Comparative Anatomy",
"Veterinary Pathology",
"Animal Nutrition",
"Zoo and Wildlife Medicine",
"Aquatic Animal Health",
"Veterinary Epidemiology",
"Animal Welfare and Ethics",
"Livestock Management",
"Poultry Science",
"Equine Science",
"Beekeeping and Apiculture",
"Wildlife Conservation",
"Animal Genetics and Breeding",
"Veterinary Immunology",
"Veterinary Pharmacology",
"Pet Medicine and Surgery",
"Emergency and Critical Care for Animals",
"Animal-Assisted Therapy",
"Exotic Animal Medicine",
"Laboratory Animal Medicine",
"Shelter Medicine and Population Control",
"Veterinary Public Health",
"One Health Approach",
"Zoonotic Diseases",
"Veterinary Surgery",
]
# Ensure you have an appropriate Wikipedia search function/library imported
for query, query1, query2 in zip(science_topics, literature_topics, list_):
# Process the first query
search_result = wp.search(query, results=500)
print(f"Query: {query}")
print(f"Results: {search_result[:5]}") # Print first 5 results for brevity
print(f"Total Results: {len(search_result)}")
print("---------------------------------------------------")
# Process the second query
search_result1 = wp.search(query1, results=500)
print(f"Query: {query1}")
print(f"Results: {search_result1[:5]}") # Print first 5 results for brevity
print(f"Total Results: {len(search_result1)}")
print("---------------------------------------------------")
# Process the third query
search_result2 = wp.search(query2, results=500)
print(f"Query: {query2}")
print(f"Results: {search_result2[:5]}") # Print first 5 results for brevity
print(f"Total Results: {len(search_result2)}")
print("---------------------------------------------------")
for title in search_result:
document = {'title': title}
try:
PagesCollection.insert_one(document)
print(f"Inserted a document successfully! {title}")
except Exception as e:
print(e)
try:
client.admin.command('ping')
print("Pinged your deployment. You successfully connected to MongoDB!")
except Exception as e:
print(e)
</code>
|
{
"filename": "Adder.ipynb",
"repository": "BDR-Pro/QuiziWiki",
"query": "transformed_from_existing",
"size": 29087,
"sha": ""
}
|
# v2_10k Immunomes Source Code .ipynb
Repository: buttelab/10kimmunomes
<font size=8 color="gray">10k Immunomes Source Code</font>
**Welcome to the 10k Immunomes Project**
This source code creates the [10k immunomes website](http://10kimmunomes.ucsf.edu/).
This `.ipynb` file is the **only** place where code is written for the 10k website. No other files that contain code. All of the other files are either images or datasets that the 10k website uses.
Below is is a summary of how the folder/files of this project are organized:
* `10k Immunomes Source Code .ipynb` - All of the source code for the 10k immunomes website
* `www` - Where the images used in the 10k site are stored
* `data` - Where the biologic data for the 10k website is stored
* `analytics` - Prefabricated javascript file used to track website usage with [google analytics](analytics.google.com)
* `other_code` - Code that is not neccesary for running the application
To get a better understanding of how this code works, it's reccomended that you first view the simpler "Template" examples inside of the `other_code` folder. You can run the "Template" files by moving them into the same file location as `10k Immunomes Source Code .ipynb` and then clicking the "web" button.
# <font color="gray">Set Up Notebook</font>
This section gets everythng ready for us to start coding. Specifically it imports all of the required packages and loads in the datasets we need of the website.
## Import Packages
Shiny and stats packages
<code>
require(shiny)
require(digest)
require(grid)
require(MASS)
require(stats)
require(shinyjs)
</code>
Plotting Packages
<code>
library(ggplot2)
library(plotly)
library(ggthemes)
</code>
## Download Everything <small>(if necessary)</small>
Here we downloald of the necessary files to run the 10k website.
### Download Website Images
<code>
if( !'www' %in% list.files() ){
print("Downloading web resource files")
dir.create("www")
download.file(url="https://storage.googleapis.com/bakar-data/10k/www/Banner.jpeg",
destfile="www/Banner.jpeg", method="wget")
download.file(url="https://storage.googleapis.com/bakar-data/10k/www/Figure_1.png",
destfile="www/Figure_1.png", method="wget")
download.file(url="https://storage.googleapis.com/bakar-data/10k/www/Table_1.jpg",
destfile="www/Table_1.jpg", method="wget")
print("Done")
}
</code>
### Download analytics files
Here we download analytics `.js` files that google uses to track the website
<code>
if( !'analytics' %in% list.files() ){
print("Downloading analytics files")
dir.create("analytics")
download.file(url="https://storage.googleapis.com/bakar-data/10k/analytics/analytics.org.js",
destfile="analytics/analytics.org.js", method="wget")
download.file(url="https://storage.googleapis.com/bakar-data/10k/analytics/analytics.ucsf.js",
destfile="analytics/analytics.ucsf.js", method="wget")
print("Done")
}
</code>
### Download Data
This downloads the immunological data files that are used to create the graphs
<code>
if( !'data' %in% list.files() ){
print("Downloading data files")
dir.create("data")
download.file(url="https://storage.googleapis.com/bakar-data/10k/data_app/newtenkdata.rdata",
destfile="data/newtenkdata.rdata", method="wget") # Kelly's 10k dataset
download.file(url="https://storage.googleapis.com/bakar-data/10k/data_app/Guinea_RNA.csv",
destfile="data/Guinea_RNA.csv", method="wget") # New Guinea RNA data
download.file(url="https://storage.googleapis.com/bakar-data/10k/data_app/png_metabolomics.725331.csv",
destfile="data/png_metabolomics.725331.csv", method="wget") # New Guinea Metabolomics dataset
print("Done")
}
</code>
## Load in Datasets and Resources
Add the `www`, `analytics`, and `data` folder to R Shiny's file path. Se that we can display images in the website.
<code>
addResourcePath("www", paste(getwd() , "/www", sep="") )
addResourcePath("analytics", paste(getwd() , "/analytics", sep="") )
addResourcePath("data", paste(getwd() , "/data", sep="") )
</code>
Add `data_raw` folder to R shiny, so that users can download all the raw datasets.
<code>
addResourcePath("data_raw", paste(getwd() , "/data_raw", sep="") )
</code>
Load in Guinea Data
<font color="red">Accidently called a Guinea dataset Gambia, fix this later</font>
<code>
gambia_rna = read.table( "data/Guinea_RNA.csv", stringsAsFactors=FALSE, header=TRUE, sep="," )
guinea_mass_spec = read.csv( "data/png_metabolomics.725331.csv", stringsAsFactors=FALSE, header=TRUE, sep="," )
</code>
Format `guinea_mass_spec` dataset so looks nicer when plotting
<code>
guinea_mass_spec[guinea_mass_spec=="M"] = "Male"
guinea_mass_spec[guinea_mass_spec=="F"] = "Female"
guinea_mass_spec[guinea_mass_spec=="DelayedGroup"] = "Delayed Group"
guinea_mass_spec[guinea_mass_spec=="VaccinatedGroup"] = "Vaccinated Group"
</code>
For newborns datasets, `gambia_rna`. We remove everyone except the the newborns who received a delayed vacciantion. I found out who was delayed by checking out the file `guinea_mass_spec`
<code>
subjects=c("P14_D0","P14_D3","P17_D0","P17_D7","P21_D0","P21_D3","P23_D0","P23_D7","P26_D0","P26_D3","P30_D0","P30_D1","P32_D0","P32_D7","P33_D0","P33_D1","P35_D0","P35_D3")
gambia_rna = gambia_rna[,c("ensembl","hgnc",subjects)]
</code>
Load in old 10k data
<code>
if( !exists("elisaAnalytes") ){
load('data/newtenkdata1.RData')
load('data/newtenkdata2.RData')
}
</code>
## Load RNA Data
We change the data so that 10k immunomes displays the hamronized rna-seq data instead of just newborns.
### Whole Blood
We load in the counts data from `whole_blood_tpm.csv`.
<code>
whole_blood_counts = read.csv( file = 'data/whole_blood_rna_tpm.csv', stringsAsFactors=FALSE )
whole_blood_subjects = read.csv( file = 'data/subjects_summary_rna_whole_blood.csv', stringsAsFactors=FALSE )
</code>
#### Code for Making Data
```python
# Create harmonized data by matching HarmonRNA output to previous harmonized data
whole_blood_counts = read.csv( file = 'harmonized_counts.csv', stringsAsFactors=FALSE ) # Load old harmonized data
whole_blood_counts2 = read.csv( file = 'data/harmony_rna_data.csv', stringsAsFactors=FALSE ) # load harmony rna TPM output
to_keep= whole_blood_counts[ whole_blood_counts$hgnc %in% whole_blood_counts2$X, 1:2 ] # put genes of harmonyRNA in same order as old dataset
to_keep2 = to_keep[order(to_keep$hgnc),]
whole_blood_counts3 = whole_blood_counts2[order(whole_blood_counts2$X),]
whole_blood_counts4 = cbind( to_keep2$ensembl , whole_blood_counts3 ) # attach ensemble column to harmonyRNA dataset
names(whole_blood_counts4)[1] = "ensembl" # Rename columns to match old dataset
names(whole_blood_counts4)[2] = "hgnc"
names_order = c() # put the columns of the new dataset in the same order as the old dataset
for( name in names(whole_blood_counts) ){
names_order= c( names_order, which( names(checkit)== name )[1] )
}
whole_blood_counts5 = whole_blood_counts4[ , names_order ]
write.csv( whole_blood_counts5, "data/harmonized_tpm.csv", row.names=FALSE) # save new dataset
```
<code>
#dim(whole_blood_counts)
#names(whole_blood_counts)
#dim(rna_counts)
#rna_counts[1:10,1:10]
#head(adjusted_matrix)
#dim(rna_counts)
#head(gambia_rna)
#dim(rna_counts)
#head(rna_counts)
#whole_blood_subjects$Subject == names(whole_blood_counts)[c(-1,-2)]
#whole_blood_counts[1:10,1:10]
</code>
We format `whole_blood_counts` so that it expresses data in terms of **TPM**, as [calculated here](https://www.rna-seqblog.com/rpkm-fpkm-and-tpm-clearly-explained/)
### PBMC
We load in the counts data from `pbmc_harmonized_counts.csv` and `subjects_summary_rna_pbmc.csv`.
<code>
pbmc_counts = read.csv( file = 'data/pbmc_rna_counts.csv', stringsAsFactors=FALSE )
pbmc_subjects = read.csv( file = 'data/subjects_summary_rna_pbmc.csv', stringsAsFactors=FALSE )
</code>
Make sure colum names and of `pbmc_counts` and rows of `pbmc_subjects` are in same order
<code>
#all(names(pbmc_counts)[-1] == pbmc_subjects$Subject)
</code>
# <font color="blue">Initialize Shiny</font>
To make this projcect more readable we I user **<font color="green">user interface</font>** and **<font color="purple">server</font>** object. I then iteratively append code to these objects a few pieces at a time.
This line creates the **<font color="green">user interface</font>**, the "front end" part of the website that everyone sees.
<code>
ui = div()
</code>
We create the **<font color="purple">server</font>** object then we add code to it one at a time in the followign sections.
<code>
server <- function(input, output, session) { }
</code>
These lines add [google analytics](https://analytics.google.com/analytics/web/) features to the website. This makes it so that you can track the users button clicks.
<font color="orange">NOTE: I think only 1 of these files is necessary, the other should probably be removed.</font>
<code>
ui = tagAppendChild(ui, tags$head(includeScript("analytics/analytics.ucsf.js")) )
ui = tagAppendChild(ui, tags$head(includeScript("analytics/analytics.org.js")) )
</code>
We add the hidden element `page` which controls which page the user is on. This is a hack I did to make R shiny have webpages like a normal website
<code>
ui = tagAppendChild(ui, hidden( selectInput('page', choices = c('home','transcriptomics','proteomics', 'immunoassays','lab'), selected = 'home', label=NULL )) )
</code>
## <font color="gray">Helper Function: callConcat</font>
<font color="blue">callConcat</font> is a function that combines R expressions. [Link to solution](https://codeday.me/en/qa/20190306/8275.html). I use it to append code to the **<font color="purple">Server Object</font>**
<code>
callConcat = function(...) {
ll <- list(...)
ll <- lapply(ll, function(X) as.list(X)[-1])
ll <- do.call("c", ll)
as.call(c(as.symbol("{"), ll))
}
</code>
# <font color="orange">Titles</font>
This is the codes that displays the website material at the top of the page. It's all the code that is above the main buttons of the website.
To understand what this section does run the **<font color="orange">Main Page</font>** section and compare it to the actual 10k website.
## <font color="orange">Main Page
<code>
tempHtml = conditionalPanel(title = "homeAbout", condition = "input.page == 'home'",
div( class="row", style="background-color: rgb(217,234,248,.5)", div( class="col-xs-12 col col-md-10 col-md-offset-1",
img(src='www/Banner.jpeg', class="img-responsive")
)),
div( class="row", div( class="jumbotron img-responsive", style="padding: .9em; background-color: rgb(250,250,250)", #rgb(225, 243, 252)
#p(" In scientific experiments it’s important to compare experimental results against a control dataset. However, sometimes it’s not possible for a lab to easily obtain data from healthy control subjects. This is especially true in immunology, the study of the immune system, where scientists may run multiple costly tests. We built 10k immunomes so that anyone can get high quality data from healthy subjects using any of the latest scientific methods. In minutes, you will have a graph ready for your paper!"),
p("In science it’s important to compare experimental results against a control dataset. However, sometimes it’s not possible obtain data from healthy control subjects. This is especially true when studying the immune system, where scientists run multiple costly tests. 10k immunomes allows anyone to obtain high quality data and create graphics using the latest scientific methods."),
p(tags$small( strong('What is 10k Immunomes?'),
'The 10,000 Immunomes Project is a reference dataset for human immunology, \r derived from over 10,000 control subjects in the',
a(href="http://www.immport.org", "NIAID ImmPort Database",inline = T, target = "_blank"),
'. Available data include flow cytometry, CyTOF, multiplex ELISA, gene expression, RNA-Seq, Mass Spectrometry, HAI titers,\r clinical lab tests, HLA type, and others. Click one of the buttons below to view visualizations of all the datasets from that type of study. More information about the website can be found in our',
a(href = 'https://www.cell.com/cell-reports/fulltext/S2211-1247(18)31451-7', 'Cell Reports Publication',inline = T, target = "_blank"),'.',
a(href = 'mailto:BISC_Helpdesk@niaid.nih.gov', 'Contact us', inline = T, target = "_blank"),
'with queries and bug reports. All code is openly available on',
a(href = 'https://github.com/pupster90/10k_Immunomes', 'Github', inline = T, target = "_blank"), "and",
a(href = 'https://hub.docker.com/r/pupster90/io', 'Docker', inline = T, target = "_blank") ) #'~Last updated 7/25/2019.'
)
))
)
ui = tagAppendChild(ui, tempHtml )
tempHtml
</code>
## <font color="orange">Graph Pages
I put all of the pages with graphs inside of one R shiny template. Which then changes depending on what button the user clicks.
First we make the title page template
<code>
tempHtml= conditionalPanel(title = "graph_page_title", condition = "input.page != 'home'",
div( class="row", div( class="jumbotron", style="padding: .9em; background-size: cover ",
div( style="display: flex;justify-content: center; position: relative; ",
h1("Graph title text" , style="text-align: center;", id = "graph_page_header"),
actionButton("homeBtn", icon = icon("home"), label="", class="btn btn-link", style=" outline: none; padding: 15px; border: 0px; background-color: transparent; font-size: 260%; ")
),
p( tags$small( id="graph_page_text" , "Graph title text" , style="text-align:center;"))
))
)
ui = tagAppendChild( ui , tempHtml )
#tempHtml
</code>
Then we add the code that changes the title page based on which button was clicked.
<code>
temp = quote({
observeEvent( input$page , {
if( input$page == 'transcriptomics' ){
html(id="graph_page_header", html= "Transcriptomics" )
html(id="graph_page_text", html= "Transcriptomics studies gene expression through the analysis of RNA molecules. The amount of RNA molecules recorded for a gene is used to determine the level of expression. This section contains data collected from bulk RNAseq and Microarrays. Until recently Microarrays were the primary method for analyzing gene expression. RNAseq is a newer technique that provides a wider dynamic range and higher sensitivity, but at a higher cost." )
}
if( input$page == 'proteomics' ){
html(id="graph_page_header", html= "Proteomics" )
html(id="graph_page_text", html= "Proteomics is the study of proteins. Proteins are molecular machines that execute the functions of a cell. This section contains CyTOF, flow cytometry, and mass spectrometry data. Flow cytometry is a popular laser-based technology to identify cells. The CyTOF and flow cytometry data contain information on relative abundance of cell phenotypes. Mass spectrometry identifies molecules through their mass-to-charge ratio, which is measured by analyzing the ‘time-of-flight’ of charged particles. Mass spectrometry data provides scientists with an in depth look at an individual’s metabolome at the time of the sample." )
}
if( input$page == 'immunoassays' ){
html(id="graph_page_header", html= "Immunoassays" )
html(id="graph_page_text", html= "Immunoassays are a useful tool for obtaining precise measurements on the concentration of molecules, bacteria, and viruses inside of a sample. This technique relies on the strong bonding affinity between an antibody and it’s corresponding antigen. Specific antibodies are chosen to ‘grab’ the particle of interest. Immunoassays can also be used to measure the relative effectiveness of an antibody on a specific virus of interest. We provide immunoassays from ELISA, Multiplex ELISA, HAI Titer, and Virus Neutralization studies." )
}
if( input$page == 'lab' ){
html(id="graph_page_header", html= "Lab Tests" )
html(id="graph_page_text", html= "Lab tests give important information about the current status of a person's metabolism including the health of essential organs, electrolyte and acid/base balance as well as levels of blood glucose and blood proteins. We provide a diverse set of lab tests on blood count, a metabolic panel, and a lipid profile. Blood Count specifies the various amounts of different cell types in the blood. Lipid profiles measure the amount of different types of lipids like cholesterol or triglycerides. Metabolic panels measure the amount to glucose and the electrolyte balance in blood.")
}
})
})
body(server) = callConcat( body(server), temp )
#if( input$page == 'rna' ){
# html(id="graph_page_header", html= "RNA-Seq" )
# html(id="graph_page_text", html= "Flow cytometry is a popular laser-based technology to analyze cells or particles. It detects and measures physical and chemical characteristics of a population of cells. In this immunophenotyping technique suspensions of living cells are stained with specific, fluorescently labeled antibodies and then analyzed with a flow cytometer. In the flow cytometer a laser beam is focused on the cell and the light scattered from it is analyzed." )
#}
#if( input$page == 'cytometry' ){
# html(id="graph_page_header", html= "Cytometry" )
# html(id="graph_page_text", html= "Proteomics is the study of proteins. Proteins are molecular machines that execute the functions of a cell. This section contains flow CyTOF, flow cytometry, and mass spectrometry data. The CyTOF and flow cytometry data provides information on relative abundance of cell phenotypes. The mass spectrometry data provides scientists with an in depth look at an individual’s metabolome at the time of the sample." )
#}
</code>
# <font color="SkyBlue">Buttons</font>
This code creates the <font color="SkyBlue">Buttons</font> used to switch between webpages.
Add <font color="SkyBlue">Buttons</font> to <font color="green">user interface</font>
<code>
tempHtml= div(
column(3, actionButton("transcriptomicsBtn", icon = icon("dna"), label="Transcriptomics", class="btn btn-success btn-lg text-center", style="width: 100%"), style="padding: 2px; "),
column(3, actionButton("proteomicsBtn", icon = icon("microscope"), label="Proteomics", class="btn-primary btn-lg text-center",style="width: 100%"), style="padding: 2px;"),
column(3, actionButton("immunoassaysBtn", icon = icon("yahoo"), label="Immunoassays", class="btn btn-warning btn-lg text-center", style="width: 100%"), style="padding: 2px;"),
column(3, actionButton("labBtn", icon = icon("vial"), label="Lab Tests", class="btn btn-danger btn-lg text-center", style="width: 100%"), style="padding: 2px; "),
p(".", style="color:white;"),
HTML("<hr>")
)
ui = tagAppendChild(ui, tempHtml )
#tempHtml
</code>
Add functions for <font color="skyblue">button clicks</font> to the <font color="purple">server</font>
<code>
temp = quote({
observeEvent(input$homeBtn, {
updateSelectInput(session, "page", selected = 'home')
})
observeEvent(input$transcriptomicsBtn, {
updateSelectInput(session, "page", selected = 'transcriptomics')
updateSelectInput(session, "dataType", selected = 'Microarray: PBMC', choices = c('Microarray: PBMC','Microarray: Whole Blood','RNAseq: T cells','RNAseq: Whole Blood', 'RNAseq: PBMC'))#, 'Cibersort: Whole Blood'))
#updateSelectInput(session, "dataType", selected = 'Gene Expression: PBMC', choices = c('Gene Expression: PBMC','Gene Expression: Whole Blood'))
})
observeEvent(input$proteomicsBtn, {
updateSelectInput(session, "page", selected = 'proteomics')
#updateSelectInput(session, "dataType", selected = 'Mass-Spec: Newborns', choices = c('CyTOF: PBMC','Flow Cytometry: Whole Blood', 'Flow Cytometry: PBMC','Mass-Spec: Newborns'))
updateSelectInput(session, "dataType", selected = 'CyTOF: PBMC', choices = c('CyTOF: PBMC','Flow Cytometry: Whole Blood', 'Flow Cytometry: PBMC','Mass Spectrometry: Newborns'))
})
observeEvent(input$immunoassaysBtn, {
updateSelectInput(session, "page", selected = 'immunoassays')
updateSelectInput(session, "dataType", selected = 'HAI Titer', choices = c('HAI Titer','ELISA', 'Multiplex ELISA', 'Virus Neutralization Titer'))
})
observeEvent(input$labBtn, {
updateSelectInput(session, "page", selected = 'lab')
updateSelectInput(session, "dataType", selected = 'Blood Count', choices = c('Blood Count','Metabolic Panel','Lipid Profile'))
})
})
body(server) = callConcat( body(server), temp )
#observeEvent(input$cytometryBtn, {
# updateSelectInput(session, "page", selected = 'cytometry')
# #updateSelectInput(session, "dataType", selected = 'CyTOF: PBMC', choices = c('CyTOF: PBMC','Flow Cytometry: Whole Blood', 'Flow Cytometry: PBMC'))
# updateSelectInput(session, "dataType", selected = 'CyTOF: PBMC', choices = c('RNAseq: T cells','RNAseq: Newborns'))
#})
</code>
# <font color="peru">Body</font>
This code displays everything on the website that is below the <font color="SkyBlue">Buttons</font>. To get a better idea of what it does uncomment the `tempHtml` line at the end of the <font color="peru">Body Home</font> section
## <font color="peru">Body Home</font>
This is the information that is displayed underneath the buttons on the home page.
<code>
tempHtml= conditionalPanel(title = "homeMain", condition = "input.page == 'home'",
div(class="jumbotron", style="background-color: white; padding: .9em",
#column(12, column(7,tags$u( h2("Made with ImmPort", class="text-center") ))),
column( 7, a(href="http://www.immport.org", img(src='www/Figure_1.png', class="img-responsive"), inline = T, target = "_blank") ),
column( 5,
p(tags$small("Data from 242 studies and 44,775 subjects was collected from the NIAID Immunology Data and Analysis Portal,",a(href="http://www.immport.org", "ImmPort", inline = T, target = "_blank"),". It includes flow cytometry, CyTOF, mRNA expression, secreted protein levels, clinical lab tests, HAI titers, HLA type, and others. We hand curated the entire contents of ImmPort to filter for normal healthy human control subjects. Each of the data types were systematically processed and harmonized. This data constitutes the largest compendium to date of cellular and molecular immune measurements on healthy normal human subjects."))
),
p(".", style="color:white; padding:0; margin:0; font-size:50%;"), # makes a vertical space,
HTML("<hr>"),
column( 5,
tags$u( h2("10,000 Subjects") ),
p(tags$small("Below is a table displaying the number of subjects in each dataset. Counts of distinct subjects for whom raw data of each type is represented in the initial release of the 10KIP. Because many subjects contributed multiple measurement types, the totals across all measurement types substantially exceed the number of distinct subjects."))
),
column( 5,
a(href="http://www.immport.org", img(src='www/Table_1.jpg', class="img-responsive"), inline = T, target = "_blank")
),
# Youtube Video
div( class="col-xs-12 col-sm-7 col-md-6", style="padding-top: 5px;",
div( class="embed-responsive embed-responsive-16by9",
HTML('<iframe width="200" height="100" src="https://www.youtube.com/embed/pwBs4J4xDOw" class="embed-responsive-item"></iframe>')
)
),
# Links for external sites and Datasets
div( class="col-xs-12 col-sm-12 col-md-6 text-center",
p(".", style="color:white"), # makes a vertical space
h3("Learn More"),
p( #tags$small(
a(href = 'http://www.immport.org/immport-open/public/home/home', 'ImmPort Homepage'), br(),
a(href = 'https://www.cell.com/cell-reports/fulltext/S2211-1247(18)31451-7', '10,000 Immunomes Paper', target = "_blank"), br(),
a(href = 'https://www.cell.com/cell-reports/fulltext/S2211-1247(18)31080-5', 'MetaCyto Cytometry Analysis Paper', target = "_blank"), br(),
a(href = 'https://bioconductor.org/packages/release/bioc/html/MetaCyto.html', 'MetaCyto Cytometry Analysis Code', target = "_blank"), br(),
a(href = 'https://www.bu.edu/jlab/wp-assets/ComBat/Abstract.html', 'ComBat Batch Correction Algorithm', target = "_blank"), br(),
a(href = 'https://storage.googleapis.com/bakar-data/10k/data_raw/Questionnaire.zip', '10k Questionnaire Dataset', target = "_blank"), br(),
a(href = 'https://storage.googleapis.com/bakar-data/10k/data_raw/hla.csv', '10k HLA Dataset', target = "_blank"), br(),
a(href = 'mailto:BISC_Helpdesk@niaid.nih.gov','Contact Us', target = "_blank")
) #)
)
),
div(
column(12,
tags$hr(),tags$hr(),
h3('Cite 10k Immunomes'),
p("Zalocusky KA, Kan MJ, Hu Z, Dunn P, Thomson E, Wiser J, Bhattacharya S, Butte AJ. The 10,000 Immunomes Project: Building a Resource for Human Immunology. Cell reports. 2019 Oct 9;25(2):513-22. PMID:30304689" )
),
column(12,
h3('REFERENCES'),
p('1) Hu Z, Jujjavarapu C, Hughey JJ, Andorf S, Lee H, Gherardini PF, Spitzer MH, et al. Meta-analysis of Cytometry Data Reveals Racial Differences in Immune Cells. Cell Reports. 2018 Jul 31;24(5):1377-88. ' ),
p('2) Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Scientific Reports. 2016 Aug 10;6(1):20686.' ),
p('3) Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostat. 2007 Jan 1;8(1):118–27. ' ),
p('4) Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003 Apr 1;4(2):249–64. ' ),
p('5) Zalocusky KA, Kan MJ, Hu Z, Dunn P, Thomson E, Wiser J, Bhattacharya S, Butte AJ. The 10,000 Immunomes Project: Building a Resource for Human Immunology. Cell reports. 2018 Oct 9;25(2):513-22. PMID:30304689' ),
p('6) Lee AH, Shannon CP, Amenyogbe N, Bennike TB, Diray-Arce J, Idoko OT, et al. Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nature Communications. 2018 March 12;10:1092.' )
),
p(".", style="color:white") # makes a vertical space
) #<- end jumbotron
) #<-- end conidtional panel
ui = tagAppendChild(ui, tempHtml )
#tempHtml
</code>
## <font color="peru">Body Graph</font>
The code below crete the <font color="peru">Page Body</font>. The is broken up into to basic sections, the **sidebar** and the **main panel**.
* **sidebar:** This is where all the widgets are that control what is graphed
* **main panel:** This small piece of code displays the graph
### <font color="peru">Sidebar
<font color="red">This code is ugly and too long. Later I should try shortening this code by consolidating similar elements in the sidebar.
<code>
sidebar = sidebarPanel( #div( class="col-sm-4",
#Select which data type to visualize
h4('Select Data Type:'),
selectInput('dataType', label = NULL,
choices = c('CyTOF: PBMC','ELISA',
'Flow Cytometry: Whole Blood', 'Flow Cytometry: PBMC',
'Microarray: PBMC','Microarray: Whole Blood',
'HAI Titer',
'Blood Count','Metabolic Panel',
'Lipid Profile','Multiplex ELISA', 'Virus Neutralization Titer',
'RNAseq: T cells', 'RNAseq: Whole Blood', 'RNAseq: PBMC', 'Mass Spectrometry: Newborns'), #'Cibersort: Whole Blood'),
selected = 'CyTOF: PBMC'
),
##Conditional panels for analyte selection
#Which drop-down menu appears will depend on the data type the user selects.
# Analyte Selection Box
conditionalPanel(condition = "input.dataType == 'Mass Spectrometry: Newborns' ",
h4('Enter Analyte Name'),
selectizeInput( 'mass_spec_analyte', label= NULL, choices= NULL,
selected='cholesterol', options= list(maxOptions=10) )
),
conditionalPanel(condition = "input.dataType == 'CyTOF: PBMC'",
h4('Select Analyte:'),
selectInput('cytof_pbmc_analyte', label = NULL, #style="padding-bottom: 0px; margin-bottom: 0px;",
choices = cytof_pbmc_analytes,
selected = 'T_cells')
),
conditionalPanel(condition = "input.dataType == 'ELISA'",
h4('Select Analyte:'),
selectInput('elisa_analyte', label = NULL,
choices = elisaAnalytes,
selected = 'CXCL10')
),
conditionalPanel(condition = "input.dataType == 'Flow Cytometry: PBMC'",
h4('Select Analyte:'),
selectInput('flow_pbmc_analyte', label = NULL,
choices = flow_pbmc_analytes,
selected = 'T_cells')
),
# Cytometry: Additional Analyte Information
conditionalPanel(condition = "input.dataType == 'CyTOF: PBMC'",
textOutput("cytof_label"), HTML("<hr>")
),
# Normalization Box
conditionalPanel( condition = "['CyTOF: PBMC','ELISA','Flow Cytometry: PBMC','Microarray: PBMC','Microarray: Whole Blood','HAI Titer','Multiplex ELISA','Blood Count','Virus Neutralization Titer'].indexOf(input.dataType) >= 0",
fluidRow(
column( 6, checkboxInput('processing', label = 'Normalized', value = T) ),
column( 6, checkboxInput('outliers', label = 'Outliers', value = T) )
)
),
# Outliers Box
#conditionalPanel( condition = "['CyTOF: PBMC','ELISA','Flow Cytometry: PBMC','Microarray: PBMC','Microarray: Whole Blood','HAI Titer','Multiplex ELISA','Blood Count','Virus Neutralization Titer'].indexOf(input.dataType) >= 0",
# checkboxInput('processing', label = 'Normalized', value = T)
#),
# Analyte Selections
conditionalPanel(condition = "input.dataType == 'Flow Cytometry: Whole Blood'",
h4('Select Analyte:'),
selectInput('flow_blood_analyte', label = NULL,
choices = flow_blood_analytes,
selected = 'T_cells')
),
conditionalPanel(condition = "input.dataType == 'Microarray: PBMC'",
h4('Enter HUGO Gene Symbol:'),
selectizeInput( 'gene_pbmc_analyte', label= NULL, choices= NULL,
selected='CD9', options= list(maxOptions=10) )
#textInput('gene_pbmc_analyte', label = NULL,
# value = 'CD9')
),
conditionalPanel(condition = "input.dataType == 'Microarray: Whole Blood'",
h4('Enter HUGO Gene Symbol:'),
selectizeInput( 'gene_blood_analyte', label= NULL, choices= NULL,
selected='CD9', options= list(maxOptions=10) )
#textInput('gene_blood_analyte', label = NULL,
# value = 'CD9')
),
conditionalPanel(condition = "input.dataType == 'HAI Titer'",
h4('Select Analyte:'),
selectInput('hai_analyte', label = NULL,
choices = haiAnalytes,
selected = haiAnalytes[1])
),
conditionalPanel(condition = "input.dataType == 'Multiplex ELISA'",
h4('Select Analyte:'),
selectInput('mbaaAnalyte', label = NULL,
choices = mbaaAnalytes,
selected = 'CXCL5')
),
conditionalPanel(condition = "input.dataType == 'Blood Count'",
h4('Select Analyte:'),
selectInput('cbcAnalyte', label = NULL,
choices = cbcAnalytes,
selected = 'HGB_g_per_dL')
),
conditionalPanel(condition = "input.dataType == 'Lipid Profile'",
h4('Select Analyte:'),
selectInput('flpAnalyte', label = NULL,
choices = flpAnalytes,
selected = flpAnalytes[1])
),
conditionalPanel(condition = "input.dataType == 'Metabolic Panel'",
h4('Select Analyte:'),
selectInput('cmpAnalyte', label = NULL,
choices = cmpAnalytes,
selected = cmpAnalytes[1])
),
conditionalPanel(condition = "input.dataType == 'Virus Neutralization Titer'",
h4('Select Analyte:'),
selectInput('vntAnalyte', label = NULL,
choices = vntAnalytes,
selected = 'Measles_Edmonston')
),
conditionalPanel(condition = "input.dataType == 'RNAseq: T cells' ",
h4('Enter HUGO Gene Symbol:'),
selectizeInput( 'T_cell_analyte', label= NULL, choices= NULL,
selected='CD9', options= list(maxOptions=10) )
#textInput('T_cell_analyte', label = NULL,
# value = 'CD9')
),
conditionalPanel(condition = "input.dataType == 'RNAseq: Whole Blood' ",
h4('Enter HUGO Gene Symbol:'),
selectizeInput( 'RNAseq_newborns', label= NULL, choices= NULL,
selected='CD9', options= list(maxOptions=10) )
#textInput('T_cell_analyte', label = NULL,
# value = 'CD9')
),
conditionalPanel(condition = "input.dataType == 'RNAseq: PBMC' ",
h4('Enter HUGO Gene Symbol:'),
selectizeInput( 'RNAseq_pbmc', label= NULL, choices= NULL,
selected='CD4', options= list(maxOptions=10) )
#textInput('T_cell_analyte', label = NULL,
# value = 'CD9')
),
conditionalPanel( condition = "input.dataType == 'Mass Spectrometry: Newborns' || input.dataType == 'RNAseq: Whole Blood' || input.dataType == 'RNAseq: PBMC'", #|| input.dataType == 'Cibersort: Whole Blood' ",
h4('Plot By:'),
radioButtons( 'newbornPlotBy', label = NULL,
#choices = c('Days of Life','Sex','Vaccination'),
choices = c('Age','Sex'), # we removed vaccination, since we are now only plotting 'delayed vaccine' group
selected = 'Age'
)
),
#Select age range of subjects
conditionalPanel(condition = "input.dataType != 'RNAseq: T cells' && input.dataType != 'RNAseq: Whole Blood' && input.dataType != 'RNAseq: PBMC' && input.dataType != 'Mass Spectrometry: Newborns'", #&& input.dataType != 'Cibersort: Whole Blood'",
h4('Age Range:'),
sliderInput("ageRange",label = NULL,
min = 0,
max = 100,
value = c(0,100),
dragRange = TRUE,
round = TRUE,
step = 1),
#Select the ethnicities of subjects
fluidRow(
column( 8,
h4('Ethnicities:'),
checkboxGroupInput('race', label = NULL,
choices = c('White', 'Black or African American','Asian','Other'),
selected = c('White', 'Black or African American','Asian','Other')
)
),
#How to color plot
column( 4 ,
h4('Plot By:'),
radioButtons('colorCode', label = NULL,
choices = c('Age & Sex','Ethnicity','Study'),
selected = 'Age & Sex'
)
)
),
fluidRow(
#Select male or female subjects
column( 12 ,
h4('Sex:'),
checkboxGroupInput("gender", label = NULL, choices = c('Female','Male'), selected = c('Female','Male'), inline=TRUE )
)
)
)
)
</code>
To make the app load quickly, we make the *"selectize inputs"* in the **<font color="orange">sidebar</font>** run using **<font color="purple">the server</font>**, as [explained here](https://shiny.rstudio.com/articles/selectize.html)
<code>
temp= quote({
updateSelectizeInput(session, 'mass_spec_analyte', choices= colnames(guinea_mass_spec)[-1:-8], server= TRUE)
updateSelectizeInput(session, 'gene_pbmc_analyte', choices= colnames(gene_pbmc)[-1:-8], server= TRUE)
updateSelectizeInput(session, 'gene_blood_analyte', choices= colnames(gene_blood)[-1:-8], server= TRUE)
updateSelectizeInput(session, 'T_cell_analyte', choices= colnames(gene_Tcell_raw)[-1:-3], server= TRUE)
updateSelectizeInput(session, 'RNAseq_newborns', choices= whole_blood_counts$hgnc, server= TRUE)
updateSelectizeInput(session, 'RNAseq_pbmc', choices= pbmc_counts$hgnc, server= TRUE)
# DEBUG
#updateSelectizeInput(session, 'RNAseq_newborns', choices= gambia_rna$hgnc, server= TRUE)
})
body(server) = callConcat( body(server), temp )
</code>
#### <font color="gray">Cleaning Code, Failed</font>
```python
sidebar = sidebarPanel(
#Select which data type to visualize
h4('Select Data Type:'),
selectInput('dataType', label = NULL,
choices = c('CyTOF: PBMC','ELISA',
'Flow Cytometry: Whole Blood', 'Flow Cytometry: PBMC',
'Gene Expression: PBMC','Gene Expression: Whole Blood',
'HAI Titer',
'Lab Tests: Blood Count','Lab Tests: Metabolic Panel',
'Lab Tests: Lipid Profile','Multiplex ELISA', 'Virus Neutralization Titer',
'RNAseq: T cells', 'Newborn RNAseq'),
selected = 'CyTOF: PBMC'
),
# Select Analyte
conditionalPanel(condition = "is.element( input.dataType, c('CyTOF: PBMC','ELISA','Flow Cytometry: Whole Blood','Flow Cytometry: PBMC','HAI Titer','Multiplex ELISA','Lab Tests: Blood Count','Lab Tests: Lipid Profile','Lab Tests: Metabolic Panel','Virus Neutralization Titer') )",
h4('Select Analyte:'),
selectInput('analyte', label = NULL, choices = cytof_pbmc_analytes, selected = 'T_cells')
),
# Normalized
conditionalPanel(condition = "is.element( input.dataType, c('CyTOF: PBMC','ELISA','Flow Cytometry: Whole Blood','Flow Cytometry: PBMC','Gene Expression: Whole Blood','HAI Titer','Multiplex ELISA','Lab Tests: Blood Count','Virus Neutralization Titer','Gene Expression: PBMC') )",
checkboxInput('processing', label = 'Normalized', value = T)
),
# Hugo Symbol
conditionalPanel(condition = "input.dataType == 'RNAseq: T cells'",
h4('Enter HUGO Gene Symbol:'),
textInput('HUGO', label = NULL, value = 'CD9')
),
#Select age range of subjects
conditionalPanel(condition = "is.element( input.dataType, c('RNAseq: T cells','Newborn RNAseq') )",
h4('Age Range:'),
sliderInput("ageRange",label = NULL,
min = 0,
max = 100,
value = c(0,100),
dragRange = TRUE,
round = TRUE,
step = 1),
#Select male or female subjects
h4('Sex:'),
checkboxGroupInput("gender", label = NULL,
choices = c('Female','Male'),
selected = c('Female','Male')),
#Select the ethnicities of subjects
h4('Ethnicities:'),
checkboxGroupInput('race', label = NULL,
choices = c('White', 'Black or African American','Asian','Other'),
selected = c('White', 'Black or African American','Asian','Other')
),
#How to color plot
h4('Plot By:'),
radioButtons('colorCode', label = NULL,
choices = c('Age & Sex','Ethnicity','Study'),
selected = 'Age & Sex'
)
)
)
```
### <font color="gray">Helper</font> <font color="blue">myDownloadButton</font>
I customized R Shiny's download button to make it look nice for the app. The code I used to do this comes [from here](https://stackoverflow.com/questions/49350509/adding-removing-icon-in-downloadbutton-and-fileinput)
<code>
myDownloadButton <- function(outputId, label = "Download", myIcon="download", class = NULL, ... ){
tags$a(id = outputId, class = paste("btn btn-default shiny-download-link", class),
href = "", target = "_blank", download = NA,
icon(myIcon) , label, ...)
}
</code>
### <font color="peru">Graph
<font color="red">Note:</font> in the future uncomment out `numSubjects` to print number of subjects.
<code>
main_panel = mainPanel( #div( class="col-sm-8",
fluidRow( div( class="col-md-offset-9 col-xs-offset-8", #col-sm-offset-8 col-lg-offset-9
tags$b( style="font-family: 'Times New Roman'; color: DarkSlateGrey; font-size: 115%", textOutput("num_subjects",inline = F) ) #
) ),
plotlyOutput("dataPlot"),
#tags$div( style = 'width:100; float:right; color:black',tags$b( textOutput("num_subjects",inline = F) ) ),
#tags$div(class = 'container2', style = 'width:100; float:right; color:black',textOutput('nSubsText', inline = F)),
#h6( id='numSubjects', class="pull-right", "Number of Subjects: " ),
h2( class="col-xs-offset-2 col-md-offset-1", "Download"),
div(
myDownloadButton('downloadPlot', label='Image', myIcon="image", class="btn btn-success"),
myDownloadButton('downloadPlotData', label='Plot Data', myIcon="file-download", class="btn btn-warning"),
tags$button( id="downloadAllData", class="btn", style="padding:0"#, # id='downloadAllData'
#HTML("<a href='https://storage.googleapis.com/bakar-data/10k/png_metabolomics.725331.csv' download ><button class='btn btn-danger'><i class='fa fa-cart-arrow-down'></i> All Data</button></a>")
)
#myDownloadButton('downloadAllData', label='All Data', myIcon="cart-arrow-down", class="btn btn-danger")
),
p( style = 'color: DarkGrey; padding: 0px;', "* All Data is dataset's raw and formatted files")
#h6( id='numSubjects', class="pull-right", "Number of Subjects: " ),
#textOutput("num_subjects")
)
</code>
Putting Graph and SideBar together
<code>
tempHtml = conditionalPanel( title="graphMain", condition = "input.page != 'home'",
sidebarLayout( sidebar, main_panel )
)
ui = tagAppendChild(ui, tempHtml )
#tempHtml
</code>
### <font color="peru">Data Explanations
Template for Data explanation text
<code>
tempHtml = conditionalPanel( title="info_text", condition = "input.page != 'home'",
div( class="jumbotron", style="background-color: white; padding: .9em;", # padding-left: .9em; padding-top: 0px;",
p( id="graph_data_info" , '..........' )
)
)
ui = tagAppendChild(ui, tempHtml )
</code>
Code that switches what template says based on selected dataset
<code>
temp = quote({
observeEvent( input$dataType , {
if( input$dataType == 'CyTOF: PBMC' ){
html(id="graph_data_info", html= "<b>CYTOF Data: </b><small>CyTOF data of healthy human blood samples were downloaded from ImmPort web portal. Every .fcs file was pared down to 5000 events. These .fcs files constitute the “raw” CyTOF:PBMC data. All data were arcsinh transformed. For CyTOF data, the formula f(x) = arcsinh (x/8) was used. Transformation and compensation were done using the preprocessing.batch function in MetaCyto (1). The cell definitions from the Human ImmunoPhenotyping Consortium (2) were used to identify 24 types of immune cells using the searchClster.batch function in MetaCyto. Specifically, each marker in each cytometry panels was bisected into positive and negative regions. Cells fulfilling the cell definitions are identified. For example, the CD14+ CD33+ CD16- (CD16- monocytes) cell subset corresponds to the cells that fall into the CD14+ region, CD33+ region and CD16- region concurrently. The proportion of each cell subsets in the PBMC were then calculated by dividing the number of cells in a subset by the total number of cells in the blood. These steps together produce the “formatted” CyTOF: PBMC data. These data were then batch-corrected with an established empirical Bayes method (3), using study accession for batch and age, sex, and race as known covariates to produce the “formatted and normalized” CyTOF: PBMC data. </small>" )
}else if( input$dataType == 'ELISA' ){
html(id="graph_data_info", html= "<b>ELISA Data: </b><small>Parsed ELISA data were downloaded from ImmPort. Analyte names were standardized to HUGO gene names where appropriate, and measurements were standardized to a common unit of measurement (pg/mL). These steps produced the “formatted” ELISA data. Because ELISA data is low-throughput, and most subjects only have measurements for one analyte, batch correction was conducted with a simple linear model for each analyte, mean correcting by study accession while accounting for age, sex, and race. These steps produced the “formatted and normalized” ELISA data.</small>" )
}else if( input$dataType %in% c('Flow Cytometry: Whole Blood','Flow Cytometry: PBMC') ){
html(id="graph_data_info", html= "<b>Cytometry Data: </b><small>Meta-analysis of Cytometry data is conducted using the MetaCyto package (1). Briefly, flow cytometry data were downloaded from ImmPort web portal. Every .fcs file was pared down to 5000 events. These .fcs files constitute the “raw” Flow Cytometry:PBMC data. Flow cytometry data from ImmPort were compensated for fluorescence spillovers using the compensation matrix supplied in each fcs file. All data from ImmPort were arcsinh transformed. For flow cytometry data, the formula f(x) = arcsinh (x/150) was used. Transformation and compensation were done using the preprocessing.batch function in MetaCyto (1). The cell definitions from the Human ImmunoPhenotyping Consortium (2) were used to identify 24 types of immune cells using the searchClster.batch function in MetaCyto. Specifically, each marker in each cytometry panels was bisected into positive and negative regions. Cells fulfilling the cell definitions are identified. For example, the CD14+ CD33+ CD16- (CD16- monocytes) cell subset corresponds to the cells that fall into the CD14+ region, CD33+ region and CD16- region concurrently. The proportion of each cell subsets in the PBMC or whole blood were then calculated by dividing the number of cells in a subset by the total number of cells in the blood. These steps together produce the “formatted” Flow Cytometry: PBMC data. Because the Flow Cytometry data are sparse, batch correction was conducted with a simple linear model for each cell type, mean correcting by study accession while accounting for age, sex, and race. These steps produced the “formatted and normalized” Flow Cytometry data.</small>")
}else if( input$dataType %in% c('Microarray: PBMC','Microarray: Whole Blood') ){
html(id="graph_data_info", html= "<b>Microarray Data: </b><small>Gene expression array data were obtained in three formats. Data in their original formats (.CEL files, series matrix files, etc) constitute the “raw” gene expression data. For data collected on Affymetrix platforms, we utilized the ReadAffy utility in the affy Bioconductor package to read in raw .CEL files. The rma utility was used to conduct Robust Multichip Average (rma) background correction (as in (4)), quantile normalization, and log2 normalization of the data. For data collected on Illumina platforms and stored in the Gene Expression Omnibus (GEO) database, we utilized the getGEO utility in the GEOquery Bioconductor package to read the expression files and the preprocessCore package to conduction rma background correction, quantile normalization, and log2 normalization of the gene expression data. Finally, for data collected on Illumina platforms but not stored in GEO, we utilized the read.ilmn utility of the limma Bioconductor package to read in the data, and the neqc function to rma background correct, quantile normalize, and log2 normalize the gene expression data. In all instances, probe IDs were converted to Entrez Gene IDs. Where multiple probes mapped to the same Entrez Gene ID, the median value across probes was used to represent the expression value of the corresponding gene. The background-corrected and normalized datasets were combined based on common Entrez IDs, missing values were imputed with a k-nearest neighbors algorithm (R package: impute, function: impute.knn) using k = 10 and default values for rowmax, colmax, and maxp. Enter Gene IDs were then converted to HUGO gene names. These steps together produced the “formatted” gene expression files. To create the “formatted and normalized” datasets, we utilized established empirical Bayes algorithm for batch correction (2), compensating for possible batch effects while maintaining potential effects of age, race, and sex across datasets.</small>")
}else if( input$dataType == 'HAI Titer' ){
html(id="graph_data_info", html= "<b>HAI Titer Data: </b><small>Parsed HAI data were downloaded from ImmPort. Names were standardized to WHO viral nomenclature where necessary. These steps produced the “formatted” HAI data. Because HAI data is low-throughput, and most subjects only have measurements for one-to-three of the viruses, batch correction was conducted with a simple linear model for each analyte, mean correcting by study accession while accounting for age, sex, and race. These steps produced the “formatted and normalized” HAI data.</small>")
}else if( input$dataType %in% c('Blood Count','Metabolic Panel','Lipid Profile') ){
html(id="graph_data_info", html= "<b>Lab Test Data: </b><small>Parsed lab test data were downloaded from ImmPort and organized into three standard panels: Complete Blood Count (CBC), Fasting Lipid Profile (FLP), and Comprehensive Metabolic Panel (CMP). Because FLP and CMP data are derived from only one study, no further standardization was required. These parsed data constitute the 'formatted' lab test data for these two types, and no 'normalized' table is available. CBC data were derived from 12 different studies. As such, names of individual tests as well as units of measurement needed to be standardized for the data to be directly comparable. For example, cells reported as thousands of cells per microliter were variously described as 'K/mi', 'K/', “cells/mm3”, “thou/mcL”, ”per”, “1000/microliter”, “10^3/mm3”, “10^3”, “1e3/uL”, “10*3/ul”, “/uL”, or “10^3 cells/uL”, and the names of assays were comparably variable. These standardization steps produced the “formatted” Lab Test: Blood Count data. These data were then batch corrected with a simple linear model for each analyte, mean correcting by study accession while accounting for age, sex, and race to produce the “formatted and normalized” CBC data.</small>")
}else if( input$dataType == 'Multiplex ELISA' ){
html(id="graph_data_info", html= "<b>Multiplex ELISA Data: </b><small>Secreted protein data measured on the multiplex ELISA platform were collected from ImmPort studies SDY22, SDY23, SDY111, SDY113, SDY180, SDY202, SDY305, SDY311, SDY312, SDY315, SDY420, SDY472, SDY478, SDY514, SDY515, SDY519, and SDY720. Data were drawn from the ImmPort parsed data tables using RMySQL or loaded into R from user-submitted unparsed data tables. Across the studies that contribute data, there are disparities in terms of the dilution of samples and units of measure in which the data are reported. We corrected for differences in dilution factor and units of measure across experiments and standardized labels associated with each protein as HUGO gene symbols. This step represents the “formatted” Multiplex ELISA data table. Compensation for batch effects was conducted using an established empirical Bayes algorithm (2), with study accession representing batch and a model matrix that included age, sex, and race of each subject. Data were log2 transformed before normalization to better fit the assumption that the data are normally distributed. The effectiveness of the log2 transform, as well as our batch correction efforts, are detailed in the manuscript associated with this resource (5). This batch-corrected data represents the “formatted and normalized” Multiplex ELISA data. </small>")
}else if( input$dataType == 'Virus Neutralization Titer' ){
html(id="graph_data_info", html= "<b>Virus Neutralization Data: </b><small>Parsed VNT data were downloaded from ImmPort. Names were standardized to WHO viral nomenclature where necessary. These steps produced the “formatted” VNT data. Because VNT data is low-throughput, and most subjects only have measurements for one-to-three of the viruses, batch correction was conducted with a simple linear model for each analyte, mean correcting by study accession while accounting for age, sex, and race. These steps produced the “formatted and normalized” VNT data.</small>")
}else if( input$dataType %in% c( 'RNAseq: Whole Blood', 'RNAseq: PBMC') ){ #, 'Cibersort: Whole Blood') ){
html(id="graph_data_info", html= "<b>RNA-Seq Whole Blood and PBMC Data: </b><small>The RNA-Seq whole blood data was downloaded from Immport (Studies: SDY1092, SDY1172, SDY1381, and SDY1412). The RNA-Seq peripheral mononucler cells comes from Immport study SDY67. The files used from Immport were formatted and normalized by the EPIC Consortium. The whole blood datasets were converted into TPM files and harmonized together using the HarmonyRNA algorithm. More information on the study can be found in this <a href=‘https://www.nature.com/articles/s41467-019-08794-x’>Nature Article</a> published by Amy H. Lee, et al. Some minor additional formatting was done for presenting data on 10k Immunomes.</small>")
}else if( input$dataType %in% c('Mass Spectrometry: Newborns') ){ #, 'Cibersort: Whole Blood') ){
html(id="graph_data_info", html= "<b>Newborn Mass Spectrometry Data: </b><small>The newborn RNA-Seq and Mass Spectrometry datasets were downloaded from Immport (Studies: SDY1256 and SDY1412). The RNAseq data comes from peripheral blood and Mass-Spec data comes from blood plasma. Phenotyping data was obtaiend by from running Cibersort on RNAseq data. The files used from Immport were formatted and normalized by the EPIC Consortium. More information on the study can be found in this <a href=‘https://www.nature.com/articles/s41467-019-08794-x’>Nature Article</a> published by Amy H. Lee, et al. Some minor additional formatting was done for presenting data on 10k Immunomes.</small>")
}else if( input$dataType == 'RNAseq: T cells' ){
html(id="graph_data_info", html= "<b>RNA-Seq T Cell Data: </b><small>The T cells' raw sequence read files were download from the Sequence Read Archive (SRA). The datasets relate to ImmPort studies SDY888 and SDY1324. The sequence reads are quantified at the gene level using Kallisto. The Transcripts per million (TPM) is used to quantify the transcription levels in T cells.</small>")
}else{
html(id="graph_data_info", html= " ")
}
})
})
body(server) = callConcat( body(server), temp )
</code>
# <font color="purple">Graph Functions</font>
This is the code that creates all of the graphs of the different datasets. This code is put inside of the <font color="purple">Server Object</font>. Three different people have added datasets 10 Immunomes, **Kelly, Zicheng, and Elliott**. I broke up the graphing portion into sections based on who created the original datasets.
## <font color="gray">Helper:</font> <font color="purple">Set analytes
This helper function is used by both **Kelly, Zicheng, and Elliott**. It sets what specific part of a dataset the viewer wishes to analyze to a varaible names `analyte`
<code>
temp= quote({
#select the analyte for plotting based on user input
analyte <- reactive({ req(switch(input$dataType, #<-- req() makes it so that the switch stops, when user hasn't made selection yet (stops errors)
"CyTOF: PBMC" = input$cytof_pbmc_analyte,
"ELISA" = input$elisa_analyte,
"Flow Cytometry: PBMC" = input$flow_pbmc_analyte,
"Flow Cytometry: Whole Blood" = input$flow_blood_analyte,
"Microarray: Whole Blood" = input$gene_blood_analyte,
"Microarray: PBMC" = input$gene_pbmc_analyte,
"Gene Set Enrichment: Whole Blood" = input$gsea_analyte,
"HAI Titer" = input$hai_analyte,
"HLA Type" = input$hla_analyte,
"Multiplex ELISA" = input$mbaaAnalyte,
"Blood Count" = input$cbcAnalyte,
"Lipid Profile" = input$flpAnalyte,
"Metabolic Panel" = input$cmpAnalyte,
"Virus Neutralization Titer" = input$vntAnalyte,
"RNAseq: T cells" = input$T_cell_analyte,
"RNAseq: Whole Blood" = input$RNAseq_newborns,
"RNAseq: PBMC" = input$RNAseq_pbmc,
#"Cibersort: Whole Blood" = input$cibersort_analyte,
"Mass Spectrometry: Newborns" = input$mass_spec_analyte
))})
})
body(server) = callConcat( body(server), temp )
</code>
### <font color="gray">Cleaning Code, Failed</font>
```python
#updateSelectInput(session, "dataType", selected = 'RNAseq: T cells', choices = c('RNAseq: T cells','Newborn RNAseq'))
tempHtml=temp= quote({
#select the analyte for plotting based on user input
analyte <- reactive({
# Get analytes of seleceted dataset and setup input box
if( input$dataType %in% c('CyTOF: PBMC','ELISA','Flow Cytometry: Whole Blood','Flow Cytometry: PBMC','HAI Titer','Multiplex ELISA','Lab Tests: Blood Count','Lab Tests: Lipid Profile','Lab Tests: Metabolic Panel','Virus Neutralization Titer') ){
choices = switch(input$dataType,
"CyTOF: PBMC" = cytof_pbmc_analytes,
"ELISA" = elisaAnalytes,
"Flow Cytometry: PBMC" = flow_pbmc_analytes,
"Flow Cytometry: Whole Blood" = flow_blood_analytes,
"HAI Titer" = haiAnalytes,
"Multiplex ELISA" = mbaaAnalytes,
"Lab Tests: Blood Count" = cbcAnalytes,
"Lab Tests: Lipid Profile" = flpAnalytes,
"Lab Tests: Metabolic Panel" = cmpAnalytes,
'Virus Neutralization Titer' = vntAnalytes
)
updateSelectInput( session, "analyte", selected = choices[1], choices = choices )
new_analyte = input$analyte
}else{ #( input.dataType %in% c('Gene Expression: PBMC','Gene Expression: Whole Blood','RNAseq: T cells','Newborn RNAseq') ){
new_analyte = input$HUGO
}
new_analyte
})
})
boedy(server) = callConcat( body(server), temp )
```
## <font color="purple">Kelly Plots
### <font color="gray">Helper: <font color="purple">Set data
<code>
kelly_data= list('Multiplex ELISA RAW'=mbaa_raw, 'Flow Cytometry: PBMC RAW'=flow_pbmc_raw, 'CyTOF: PBMC RAW'=cytof_pbmc_raw, "ELISA RAW"=elisa_raw,
"Microarray: Whole Blood RAW"=gene_blood_raw, "Microarray: PBMC RAW"=gene_pbmc_raw, "HAI Titer"=hai_raw, "Blood Count RAW"=cbc_raw,
'Virus Neutralization Titer RAW'=vnt_raw, "CyTOF: PBMC"=cytof_pbmc, "ELISA"=elisa, "Flow Cytometry: PBMC"=flow_pbmc, "Blood Count"=cbc,
"Flow Cytometry: Whole Blood"=flow_blood_raw, "Microarray: Whole Blood"=gene_blood, 'Virus Neutralization Titer' = vnt,
"Microarray: PBMC"=gene_pbmc, "HAI Titer"=hai, "HLA Type"=hla, "Multiplex ELISA"=mbaa, "Lipid Profile"=flp, "Metabolic Panel" = cmp #,
)
</code>
### <font color="gray">Helper: <font color="purple">Titles and Y-axis labels
<code>
temp= quote({
# Generate a Title
title <- reactive({paste(gsub('_',' ',analyte()), 'by Subject')})
# Generate Labels
ylab <- reactive({
switch( input$dataType ,
"CyTOF: PBMC" = paste(gsub('_',' ',analyte()),'(percent)'),
"ELISA"= paste(analyte(), '(Concentration in pg/mL)'),
"Flow Cytometry: PBMC" = paste(gsub('_',' ',analyte()),'(percent)'),
"Flow Cytometry: Whole Blood" = paste(gsub('_',' ',analyte()), '(percent)'),
"Microarray: Whole Blood" = paste(analyte(), '(Expression)'),
"Microarray: PBMC" = paste(analyte(), '(Expression)'),
"Gene Set Enrichment: Whole Blood" = paste(analyte()),
"HAI Titer" = paste(analyte(), '(HAI Titer)'),
"Multiplex ELISA" = paste(analyte(), '(Concentration in pg/mL)'),
"Blood Count" = analyte(),
"Lipid Profile" = analyte(),
"Metabolic Panel" = analyte(),
"Virus Neutralization Titer" = paste(analyte(), '(Virus Neutralization Titer)'),
"RNAseq: T cells" = paste(analyte(), '(Expression)'),
"RNAseq: Newborns" = paste(analyte(), '(Expression)'),
#"Cibersort: Whole Blood" = paste(analyte(), '(Expression)'),
"Mass Spectrometry: Newborns" = paste(analyte(), '(Expression)')
)
})
})
body(server) = callConcat( body(server), temp )
</code>
### <font color="gray">Helper: <font color="purple">Cytometry Data's Extra Infro
This is additional information that is displayed in the cytometry sidebar when the user is looking at the "CyTOF: PBMC" dataset
<code>
cyto_labels= list('B_cells'="CD19+,CD3- or CD19+,CD20+", 'CD16_neg_monocytes'="CD14+,CD33+,CD16-", 'CD16_pos_monocytes'="CD14+,CD33+,CD16+",
'CD4_T_cells'="CD3+,CD4+", 'CD8_T_cells'="CD3+,CD8+", 'Central_Memory_CD4_T_cells'="CD3+,CD4+,CCR7+,CD45RA-",
'Central_Memory_CD8_T_cells'="CD3+,CD8+,CCR7+,CD45RA-", 'Effector_CD4_T_cells'="CD3+,CD4+,CCR7-,CD45RA+",
'Effector_CD8_T_cells'="CD3+,CD8+,CCR7-,CD45RA+", 'Effector_Memory_CD4_T_cells'="CD3+,CD4+,CCR7-,CD45RA-",
'Effector_Memory_CD8_T_cells'="CD3+,CD8+,CCR7-,CD45RA-", 'Gamma_Delta_T_cells'="TCRgd+,CD3+", 'Lymphocytes'="CD14-,CD33-",
'Memory_B_cells'="CD3-,CD19+,CD20+,CD24+,CD38-", 'Monocytes'="CD14+,CD33+", 'Naive_B_cells'="CD3-,CD19+,CD20+,CD24-,CD38+",
'Naive_CD4_T_cells'="CD3+,CD4+,CCR7+,CD45RA+", 'Naive_CD8_T_cells'="CD3+,CD8+,CCR7+,CD45RA+", 'NK_cells'="CD3-,CD16+,CD56+",
'NKT_cells'="CD3+,CD56+", 'Plasmablasts'="CD3-,CD20-,CD27+,CD38+", 'T_cells'="CD3+",
'Transitional_B_cells'="CD3-,CD19+,CD20+,CD24+,CD38+", 'Tregs'="CD3+,CD4+,CD25+,CD127- or CD3+,CD4+,CD25+,FOXP3+"
)
</code>
### <font color="purple">Kelly Main Functions
We write the function `kellyData` which gets the dataset to plot
<code>
temp= quote({
kellyData <- reactive({
# Get dataset from kelly_data dictionary
data_name = input$dataType
if( input$processing == F && input$dataType %in% c('CyTOF: PBMC','ELISA','Flow Cytometry: PBMC','Microarray: PBMC','Microarray: Whole Blood','HAI Titer','Multiplex ELISA','Blood Count','Virus Neutralization Titer') ){
data_name = paste(data_name, "RAW" )
}
data = kelly_data[[data_name]]
# Format data for graphing
data = data[ data$age > min(input$ageRange) & data$age < max(input$ageRange) & data$gender %in% input$gender & data$race %in% input$race ,]
data= data[ !is.na( data[,analyte()] ), ]
# Remove outliers
if( input$outliers == F ){
vals= data[,analyte()]
data= data[ abs(vals- mean(vals)) < 2*sd(vals) , ]
}
kelly_data = data.frame( Age=data[,'age'], Sex=as.factor(data[,'gender']), Value=round(data[,analyte()],2), Study=factor(data[,'study_accession']), Race=data[,'race'] )
kelly_data
})
})
body(server) = callConcat( body(server), temp )
</code>
We write the funciton `kellyPlot` that creates the plot
<code>
temp= quote({
kellyPlot <- reactive({
to_graph = kellyData()
# Decide if graph on log scale
scaleChoice = 'identity'
if( input$dataType %in% c('Microarray: Whole Blood','Microarray: PBMC','Multiplex ELISA','RNAseq: T cells') ){
scaleChoice = 'log10'
}
# If cytof dataset, display analyte label
if( input$dataType == 'CyTOF: PBMC' ){
output$cytof_label= renderText({ paste("* Defined by", cyto_labels[[analyte()]] ) }) #"*", analyte(),
}
# Plot Output depending on selected "plot by"
if( input$colorCode == 'Age & Sex' ){
p <- ggplot(data=to_graph, aes(x=Age, y=Value, color=Sex, race=Race, study=Study ) ) + geom_point(alpha=.6) + theme_gdocs() +
labs(x = 'Subject Ages', y = ylab(), title = title() ) + scale_y_continuous(trans=scaleChoice) +
stat_smooth( mapping=aes(x=Age, y=Value, color=Sex), inherit.aes=FALSE, se=FALSE )
#p + stat_smooth( data=to_graph, mapping=aes(x=Age, y=Value, color=Sex), se=FALSE ) #stat_smooth(method=loess, data = to_graph[,c("Age","Value")] )
}else if( input$colorCode == 'Study' ){
p <- ggplot(data= to_graph, aes(x=Age, y=Value, sex=Sex, race=Race, color=Study) ) + geom_point(alpha=.6) + theme_gdocs() +
labs(x = 'Subject Ages', y = ylab(), title = title() ) + scale_y_continuous(trans=scaleChoice) +
stat_smooth( mapping=aes(x=Age, y=Value, color=Study), inherit.aes=FALSE, se=FALSE )
#p + scale_y_continuous(trans=scaleChoice) + stat_smooth(method=loess,data = dataBind(),aes(fill = factor(dataBind()[,'study_accession'])))+
}else{ # Plot by Ethnicity
par(las=1, bty='l', lwd=2, family = 'sans',cex.axis=1.25, cex.lab=1.25, cex.main=1.75)
p <- ggplot( data= to_graph, aes(x=Race, y= Value, fill=Race, sex=Sex, study=Study) ) +
geom_jitter( width=0.15, alpha=0.8, stroke=0, size=2 ) + #height = 0, opacity=.5
geom_violin( inherit.aes=FALSE, mapping=aes(x=Race, y= Value, fill=Race), trim= FALSE, alpha=0.5, show.legend=FALSE ) + # inherit.aes=T, colour=NA,
#geom_jitter( data=to_graph, aes(x=Race, y= Value, fill=Race, sex=Sex, study=Study), width=0.15, alpha=0.8, stroke=0, size=2 ) + #height = 0, opacity=.5
labs(x = 'Race', y = ylab(), title = title() ) +
theme_gdocs()
ggplotly(p, tooltip = c("Value", "density", "Sex", "Study") )
}
})
})
body(server) = callConcat( body(server), temp )
</code>
## <font color="purple">Zicheng Plot
We write the function `zichengData` which gets the dataset to plot
<code>
temp= quote({
zichengData <- reactive({
data.frame( Cell= gsub(" CD3.*","",gene_Tcell_raw[,'CellType']) , Value= gene_Tcell_raw[,analyte()] )
})
})
body(server) = callConcat( body(server), temp )
</code>
We write the funciton `zichengPlot` that creates the plot
<code>
temp= quote({
zichengPlot <- reactive({
# Create dataset
to_graph= zichengData()
# Create Plot
par(las=1, bty='l', lwd=2, family = 'sans', cex.axis=1.25, cex.lab=1.25, cex.main=1.75) # Some basic style formating
p <- ggplot(data= to_graph, aes(x=Cell, y =Value, fill=Cell) ) +
geom_jitter(width = 0.15, alpha = 0.75 , stroke = .3, size=2 ) + #height = 0, opacity=.5
geom_violin( trim= FALSE, alpha = 0.5, inherit.aes = T, show.legend=FALSE ) + # colour=NA,
labs( y=paste(analyte(),' Expression'), title=paste( analyte()," by Cell") )+ #paste(analyte(), '(Expression)')
theme_gdocs()
ggplotly(p, tooltip = c("Value", "density") ) # , "Sex", "Subject"
})
})
body(server) = callConcat( body(server), temp )
</code>
## <font color="purple">Elliott Plots
We write the function `elliottData` which gets the dataset to plot
<code>
#head(pbmc_subjects)
</code>
<code>
#pbmc_counts[1,1:10]
</code>
<code>
temp= quote({
elliottData <- reactive({
# Create Whole Blood RNA Data
if( input$dataType == 'RNAseq: Whole Blood' ){
num = which(whole_blood_counts$hgnc==analyte() ) # gambia_rna$hgnc
gene_name = whole_blood_counts[num,2]
to_graph = whole_blood_subjects
a_row = whole_blood_counts[num,3:dim(whole_blood_counts)[2]]
to_graph$Expression = as.numeric(a_row)
}
# Create PBMC RNA Data
else if( input$dataType == 'RNAseq: PBMC' ){
num = which(pbmc_counts$hgnc==analyte() )
gene_name = pbmc_counts[num,1]
to_graph = pbmc_subjects
a_row = pbmc_counts[num,2:dim(pbmc_counts)[2]]
to_graph$Expression = as.numeric(a_row)
}
# Create Mass Spec Data
#else if( input$dataType == "Mass Spectrometry: Newborns" ){
else{
col_num = which( colnames( guinea_mass_spec ) == analyte() )[1] # <- get column number of analyte
to_graph= data.frame( Expression= round(guinea_mass_spec[,col_num],3) , Group= as.factor( guinea_mass_spec$DAY ), Sex=guinea_mass_spec$SEX, Vaccination=guinea_mass_spec$TREATMENT )
to_graph$Day = guinea_mass_spec$DAY
to_graph$DayPlot = guinea_mass_spec$DAY + rnorm( dim(guinea_mass_spec)[1] , mean = 0, sd = .15 )
to_graph$Subject = guinea_mass_spec$SUBJECT
}
to_graph # <- return dataset
})
})
body(server) = callConcat( body(server), temp )
</code>
We write the funciton `elliottPlot` that creates the plot
<code>
temp= quote({
elliottPlot <- reactive({
to_graph = elliottData() # <- Get dataset
# For when the data is NOT RNAseq
if( input$dataType != 'RNAseq: Whole Blood' & input$dataType != 'RNAseq: PBMC' ){
# Decide how to plot data based on "newbornPlotBy" input value
main_plot = switch( input$newbornPlotBy ,
'Age' = ggplot( to_graph, aes(x=DayPlot, day=Day, y=Expression, color=Group, sex=Sex, subject=Subject ) ), #status=Vaccination,
'Sex'= ggplot( to_graph, aes(x=DayPlot, day=Day, y=Expression, color=Sex, status=Vaccination, subject=Subject ) ) #,
#'Vaccination' = ggplot( to_graph, aes(x=DayPlot, day=Day, y=Expression, color=Vaccination, sex=Sex, subject=Subject ) )
)
my_ylab = switch( input$dataType ,
'RNAseq: Newborns' = "Gene Expression (Transcripts per Million)",
"Mass Spectrometry: Newborns" = "Molecule Expression",
"Phenotype Expression"
)
p = main_plot + geom_point(alpha=.7) + theme_gdocs() + labs(x='Days Alive', y=my_ylab, title=paste( analyte()," Expression in Newborns") )
ggplotly( p , tooltip = c("Day", "Expression", "Subject", "Sex" ) ) #, "Vaccination"
}else{ # For when it is RNAseq Data
# Change code so that whole blood study SDY1092 ages are displayed as NA
to_graph$age = to_graph$Age
if( input$dataType == 'RNAseq: Whole Blood' ){
to_graph$Age = round(to_graph$Age, digits=1)
to_graph$Expression = round(to_graph$Expression, digits=1)
to_graph$Age[ to_graph$Study=="SDY1092" ] = NA
}
main_plot = switch( input$newbornPlotBy ,
'Age' = ggplot( to_graph, aes(x=age, y=Expression, color=Study, sex=Sex, subject=Subject, Age=Age ) ), #status=Vaccination,
'Sex' = ggplot( to_graph, aes(x=age, y=Expression, color=Sex, study=Study, subject=Subject, Age=Age ) ) #,
#'Vaccination' = ggplot( to_graph, aes(x=DayPlot, day=Day, y=Expression, color=Vaccination, sex=Sex, subject=Subject ) )
)
my_ylab= "Transcripts per Million"
p = main_plot + geom_point(alpha=.7) + theme_gdocs() + labs(x='Age', y=my_ylab, title=paste( analyte()," Expression") )
ggplotly( p , tooltip = c("Age", "Expression","Study","Sex", "Subject" ) ) #, "Vaccination"
}
})
})
body(server) = callConcat( body(server), temp )
</code>
## <font color="purple">Render Plot
Based on whoever's dataset (Kelly, Zicheng, or Elliott) we are looking at, we create the graph of the dataset the user want to see. We also print the number of subjects currently being graph. This graph is rendered using the [R Plotly package](https://plot.ly/r/) so that it contains a bunch of cool interactive features.
<code>
temp= quote({
plotInput <- reactive({
if( input$dataType %in% names(kelly_data) ){
p <- kellyPlot()
my_data = kellyData()
}else if( input$dataType == 'RNAseq: T cells' ){
p <- zichengPlot()
my_data = zichengData()
}else{ # these plots are: "RNAseq: Newborns" "Cibersort: Whole Blood" "Mass Spectrometry: Newborns"
p <- elliottPlot()
my_data = elliottData()
}
output$num_subjects = renderText({ paste( dim(my_data)[1]," Subjects" ) })
p
}) #<-- plotInput end
output$dataPlot <- renderPlotly({ plotInput() })
}) #<-- Quote end
body(server) = callConcat( body(server), temp )
</code>
# <font color="green">Download Data</font>
The functions below run when one of the "Download" buttons. A user can download an image, a cleaned up version of the dataset, or all of the raw data
## <font color="green">Download Image</font> <font color="red">Fix Glitches
The code that runs when "image" is clicked
<font color="red">This code currently doesn't work for every graph. Figure out why and fix it.
<code>
temp= quote({
#Handle 'Download Plot' button on Visualize page
output$downloadPlot <- downloadHandler(
filename = function() {
paste('10KImmunomes', input$dataType, analyte(), '.pdf', sep='_')
#paste('10KImmunomes', input$dataType, '.pdf', sep='_')
},
content = function(file) {
pdf(file, width = 8, height = 5)
print(plotInput())
dev.off()
}
)
}) #<-- Quote end
body(server) = callConcat( body(server), temp )
</code>
## <font color="green">Download Plot Data</font>
The code that runs when "Plot Data" is clicked
<code>
temp= quote({
output$downloadPlotData <- downloadHandler(
# Write File Name
filename = function() { paste('10KImmunomes', input$dataType, analyte(), '.csv', sep='_') }, # Sys.Date() <-- can add if u like
# Create File
content = function(file) {
# Get Dataset for file
if( input$dataType %in% names(kelly_data) ){
dataPlot <- kellyData()
}else if( input$dataType == 'RNAseq: T cells' ){
dataPlot <- zichengData()
}else{ # these plots are: "RNAseq: Newborns" "Cibersort: Whole Blood" "Mass Spectrometry: Newborns"
dataPlot <- elliottData()
}
write.csv( dataPlot, file, row.names = F) # Write File
# Get Dataset for file
#dataPlot= switch( input$dataType,
# 'RNAseq: T cells' = cbind( data()[,2:3] , data()[, analyte()] ) ,
# "RNAseq: Newborns" = data()[ which(data()$hgnc==analyte()) , ] ,
# "Mass Spectrometry: Newborns"= cbind( data()[,4:8] , data()[, analyte()] ) ,
# "Cibersort: Whole Blood"= cbind( data()[,4:8] , data()[, analyte()] ) ,
# cbind( data()[,2:5] , data()[, analyte()] ) # <-- All Other Data
# )
}
)
}) #<-- Quote end
body(server) = callConcat( body(server), temp )
</code>
## <font color="green">Download All Data</font>
The code that runs when "All Data" is clicked
<code>
temp= quote({
observeEvent( input$dataType , {
# Get the path to where the data will be downloaded from
file_path = switch(input$dataType,
"CyTOF: PBMC" = "data_raw/CyTOF_PBMC.zip",
"ELISA" = "data_raw/ELISA.zip",
"Flow Cytometry: PBMC" = "data_raw/Flow_Cytometry_PBMC.zip",
"Flow Cytometry: Whole Blood" = "data_raw/Flow_Cytometry_Whole_Blood.zip",
"Microarray: Whole Blood" = "data_raw/Gene_Expression_Whole_Blood_formatted.zip",
"Microarray: PBMC" = "data_raw/Gene_Expression_PBMC.zip", #"Gene Set Enrichment: Whole Blood" = input$gsea_analyte,
"HAI Titer" = "data_raw/HAI_Titer.zip",
"HLA Type" = "data_raw/hla.csv",
"Multiplex ELISA" = "data_raw/Multiplex_ELISA.zip",
"Blood Count" = "data_raw/Lab_Tests_Blood_Count.zip",
"Lipid Profile" = "data_raw/lab_test_fasting_lipid_profile.csv",
"Metabolic Panel" = "data_raw/lab_test_comprehensive_metabolic_panel.csv",
"Virus Neutralization Titer" = "data_raw/Virus_Neutralization_Titer.zip",
"RNAseq: T cells" = "data_raw/gene_Tcells_formatted.csv",
"RNAseq: Whole Blood" = "data/whole_blood_rna_tpm.csv",
"RNAseq: PBMC" = "data/pbmc_rna_counts.csv",
"Mass Spectrometry: Newborns" = "data/png_metabolomics.725331.csv",
)
# Change the download button's href to the download path
html_pre = "<a href='"
html_post= "' download ><button class='btn btn-danger'><i class='fa fa-cart-arrow-down'></i> All Data</button></a>"
html( id='downloadAllData', html=paste( html_pre, file_path, html_post, sep="" ) )
}) #<-- observeEvent end
}) #<-- Quote end
body(server) = callConcat( body(server), temp )
</code>
# <font color="blue">Run Shiny</font>
We change the following options so that <font color="blue">RShiny</font> runs at an exposed port and on the computer.
<code>
options(shiny.port = 8888)
options(shiny.host = "0.0.0.0")
</code>
We run the app!
<code>
shinyApp(ui = fluidPage( useShinyjs(), style='margin-left:5px; margin-right:5px', ui ), server = server)
</code>
# <font color="brown">Scratch Paper</font>
This is where I store all of the code that is currenly not being used by the website, but that might be useful in the future.
I converted the code to markdown cells so that it doesn't cause issues when clicking `run all` code cells. Turn the cells back to code cells to see what they do.
## Questionnaire Analysis
Non visualized data names
``` python
hla <- read.csv('./allDataForApp/Formatted Files/hla.csv', row.names = NULL)
questions <- read.csv('./allDataForApp/Formatted Files/questionnaire.csv', row.names = NULL)
questions_dict <- read.csv('./allDataForApp/Formatted Files/questionnaire_dictionary.csv',row.names = NULL)
```
print( questions_dict )
questions
questions[1,]
print(dim(hla))
hla
Checking to see if locus name column is unqiue
length( as.character( hla$locus_name ) )
unique( as.character( hla$locus_name ) )
## Basic ui and server
ui <- basicPage(
plotlyOutput("dataPlot")
)
server <- function(input, output) {
output$dataPlot <- renderPlotly({ # plot(x, sin(x)) })
ggplot( to_graph, aes(x=X, y=counts, color=groups) ) +
geom_point() +
labs(x='Days Alive', y="Gene Expression (counts)", title=paste(gene_name," Expression in Newborns"), fill = NULL, colour = NULL ) +
theme_gdocs()
})
}
## <font color="purple">Maybe Useful-</font> ShinyJS
The following code can be added to the notebook to add javascript and CSS features to the Shiny application.
``` python
jscode <- "
shinyjs.disableTab = function(name) {
}
"
css <- "
.nav li a.disabled {
background-color: #aaa !important;
color: #333 !important;
cursor: not-allowed !important;
border-color: #aaa !important;
}"
```
After the code is written put a line of code like the one below in the <font color="green">ui</font>
``` python
extendShinyjs(text = jscode)
```
## Maybe Useful: Boostrap Page
``` python
ui = bootstrapPage( h1("howdy Dowdy") )
```
|
{
"filename": "v2_10k Immunomes Source Code .ipynb",
"repository": "buttelab/10kimmunomes",
"query": "transformed_from_existing",
"size": 120670,
"sha": ""
}
|
# Code.ipynb
Repository: wozniakw2002/WB-2024
<code>
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import networkx as nx
import random
import warnings
warnings.filterwarnings('ignore')
# from points_io import save_points_as_pdb
# pd.options.mode.copy_on_write = True
</code>
<code>
data = pd.read_table('data/GSM1173493_cell-1.txt')
chrom1_data = data[((data["chrom1"] == '15') & (data["chrom2"] == '15'))]
chrom1_data['coord1_bin'] = pd.cut(chrom1_data['coord1'], bins=50, labels=False)
chrom1_data['coord2_bin'] = pd.cut(chrom1_data['coord2'], bins=50, labels=False)
heatmap_data_chrom1 = chrom1_data.pivot_table(index='coord1_bin', columns='coord2_bin', aggfunc='size', fill_value=0)
plt.figure(figsize=(8, 6))
sns.heatmap(heatmap_data_chrom1, cmap='YlOrRd')
plt.title('Heatmap of Hi-C interactions between Chromosome 1')
plt.xlabel('Genomic coordinate bin')
plt.ylabel('Genomic coordinate bin')
plt.show()
</code>
## Checkpoint 2
<code>
data = pd.read_table('data/GSM1173493_cell-1.txt')
chrom1_data = data[((data["chrom1"] == '15') & (data["chrom2"] == '15'))]
chrom1_data['coord1_bin'] = pd.cut(chrom1_data['coord1'], bins=150, labels=False)
chrom1_data['coord2_bin'] = pd.cut(chrom1_data['coord2'], bins=150, labels=False)
heatmap_data_chrom1 = chrom1_data.pivot_table(index='coord1_bin', columns='coord2_bin', aggfunc='size', fill_value=0)
coord1_nonzero_indices = []
coord2_nonzero_indices = []
for coord1_index in heatmap_data_chrom1.index:
for coord2_index in heatmap_data_chrom1.columns:
if heatmap_data_chrom1.loc[coord1_index, coord2_index] > 0:
coord1_nonzero_indices.append(coord1_index)
coord2_nonzero_indices.append(coord2_index)
</code>
<code>
def createGraph(coord1, coord2):
G = nx.cubical_graph()
for i in range(len(coord1)):
if coord1[i] == coord2[i]:
continue
G.add_edge(coord1[i], coord2[i])
pos = nx.spring_layout(G, dim=3)
return G, pos
</code>
<code>
G, pos = createGraph(coord1_nonzero_indices, coord2_nonzero_indices)
</code>
<code>
def random_walk(G, pos,n=1000):
matrix = np.empty((0, 3))
node = random.choice(list(G.nodes()))
coord = pos[node]
matrix = np.vstack([matrix, np.array(coord)])
for _ in range(n):
node = random.choice(list(G.neighbors(node)))
coord = pos[node]
matrix = np.vstack([matrix, np.array(coord)])
save_points_as_pdb(matrix, pdb_file_name='points.pdb')
</code>
<code>
random_walk(G, pos)
</code>
<code>
def generate_self_avoiding_walk(max_steps, grid):
walk = [(0, 0, 0)]
visited = set([(0, 0, 0)])
moves = [(0, 0, 1), (0, 0, -1), (0, 1, 0), (0, -1, 0), (1, 0, 0), (-1, 0, 0)]
for _ in range(max_steps):
dx, dy, dz = random.choice(moves)
new_pos = (walk[-1][0] + dx, walk[-1][1] + dy, walk[-1][2] + dz)
if any(abs(i) > grid for i in new_pos):
continue
if new_pos not in visited:
walk.append(new_pos)
visited.add(new_pos)
else:
continue
return walk
</code>
<code>
generate_self_avoiding_walk(10, 7)
</code>
|
{
"filename": "Code.ipynb",
"repository": "wozniakw2002/WB-2024",
"query": "transformed_from_existing",
"size": 59598,
"sha": ""
}
|
# Plant_Pathogen_Atlas_imputation_part0.ipynb
Repository: amonell/Spatial
<code>
library(Seurat)
library(Matrix)
</code>
<code>
scrnaseq_r <- readRDS('../../data/AvrRpt2_alone2.rds')
</code>
<code>
dior::write_h5(scrnaseq_r, file="../../data/AvrRpt2_alone2.h5", object.type = 'seurat')
</code>
<code>
DefaultAssay(scrnaseq_r) <- "atacRNA_400bp"
</code>
<code>
dior::write_h5(scrnaseq_r, file="../../data/AvrRpt2_alone2_atac.h5", object.type = 'seurat', assay.name = "atacRNA_400bp")
</code>
<code>
# Specify the file path where you want to save the CSV file
file_path <- "../../data/AvrRpt2_alone2_chromvar.csv"
# Write the data table to a CSV file
write.csv(scrnaseq_r@assays$chromvar$data, file = file_path)
</code>
|
{
"filename": "Plant_Pathogen_Atlas_imputation_part0.ipynb",
"repository": "amonell/Spatial",
"query": "transformed_from_existing",
"size": 2295,
"sha": ""
}
|
# 251124sepsis Panels.ipynb
Repository: TanyaJohary/sepsis-diagnosis
## Pathogen-based panels:
The article "In vitro diagnosis of sepsis: a review" focuses on the advancements and challenges in diagnosing sepsis through in vitro diagnostic methods. It addresses the limitations of traditional culture-based approaches and explores molecular diagnostic techniques that improve sensitivity, specificity, and rapidity in identifying sepsis-causing pathogens. Below is a comprehensive summary:
1. Sepsis
Sepsis is a systemic inflammatory response syndrome (SIRS) caused by infections, leading to severe sepsis or septic shock in critical cases.
It affects around 2% of hospitalized patients and has high mortality rates:
25–30% in severe sepsis.
40–70% in septic shock.
2. Challenges in Sepsis Diagnosis
Blood Culture (BC):Still the "gold standard" for identifying bloodstream infections.
Limitations:
Low sensitivity (up to 50% false negatives).
Long turnaround time (12–72 hours).
High contamination rate (~3%).
Poor performance in detecting fastidious, unculturable, or slow-growing pathogens.
Need for Molecular Diagnostics:Rapid and sensitive molecular techniques are essential to reduce mortality and improve patient outcomes.
3. Molecular Techniques for Sepsis Diagnosis
A. PCR-Based Assays
Prove-it™ Sepsis (Mobidiag):Multiplex PCR with microarray.
Detects 83 pathogens (Gram-positive, Gram-negative, fungi) and antibiotic resistance markers (e.g., mecA).
Turnaround time: 3 hours.
Sensitivity: 94.7%; Specificity: 98.8%.
LightCycler® SeptiFast:Real-time PCR detecting 25 pathogens and resistance markers (mecA).
Turnaround time: 6 hours.
Sensitivity: 79–91%; Specificity: 88–96%.
SepsiTest®:Broad-range PCR targeting 16S/18S rRNA genes.
Detects 345+ pathogens.
Turnaround time: 8–12 hours.
Sensitivity: 87%; Specificity: 86%.
B. Mass Spectrometry-Based Approaches
MALDI-TOF MS:Identifies bacteria and fungi by their proteomic profiles.
Turnaround time: 1–2 hours.
Sensitivity: 76–98%; Specificity: ~96%.
Limitations: Reduced sensitivity for Gram-positive bacteria.
PCR/ESI-MS (Abbott Molecular):Combines PCR with electrospray ionization mass spectrometry.
Detects 800+ pathogens and resistance markers (mecA, vanA/B).
Turnaround time: 6 hours.
Sensitivity: 83%; Specificity: 94%.
C. Fluorescent Hybridization Assays
PNA-FISH®:Peptide nucleic acid probes target ribosomal RNA.
Turnaround time: 2.5–3 hours.
Sensitivity and Specificity: >98%.
D. Emerging Technologies
Next-Generation Sequencing (NGS):High-throughput sequencing to detect pathogens and resistance markers.
Challenges include cost, sensitivity to low pathogen loads, and vast background human DNA.
Turnaround time: 2–3 hours (for advanced platforms).
4. Biomarkers for Sepsis
common biomarkers for sepsis:Acute-Phase Proteins:
**C-reactive protein (CRP)**: High sensitivity but limited specificity.
**Procalcitonin (PCT)**: Reliable for bacterial infections; increases within 4–8 hours of infection onset.
Cytokines:**IL-6, IL-8**: Indicators of inflammation.
Complement factors **(C3a, C5a, cC5aR)**: Early markers for sepsis severity.
5. Strengths and Limitations of Molecular Diagnostics
Strengths:Faster than traditional methods (e.g., BC).
Higher sensitivity for fastidious or non-culturable pathogens.
Simultaneous detection of multiple pathogens and resistance markers.
Limitations:High cost and need for specialized equipment.
Limited ability to assess antimicrobial susceptibility directly.
Some techniques (e.g., PCR) are prone to contamination and false positives.
6. Clinical Implications
Early diagnosis of sepsis can significantly improve patient survival.
Molecular diagnostics are complementary to traditional BC, offering rapid initial identification but often requiring BC for susceptibility testing.
A combination of multiplex PCR, NGS, and proteomics represents the future of sepsis diagnostics.
## The Pediatric Sepsis Biomarker Risk Model (PERSEVERE)
is a prognostic tool developed to estimate mortality risk and assess illness severity in children with septic shock. It utilizes a panel of biomarkers measured from blood samples obtained within the first 24 hours of clinical presentation.
Biomarkers in the PERSEVERE Panel:
The original PERSEVERE model comprises five protein biomarkers:
**C-C motif chemokine ligand 3 (CCL3)**: A chemokine involved in immune responses, particularly in attracting immune cells to sites of infection.
**Interleukin-8 (IL-8)**: A cytokine that plays a role in the inflammatory response by attracting neutrophils to infection sites.
**Heat shock protein 70 kDa 1B (HSPA1B)**: A stress-induced protein that assists in protein folding and protection under stress conditions.
**Granzyme B (GZMB)**: An enzyme released by cytotoxic T cells and natural killer cells to induce apoptosis in target cells.
**Matrix metallopeptidase 8 (MMP-8)**: An enzyme involved in the breakdown of extracellular matrix components, playing a role in tissue remodeling and inflammation.
## Gene Expression Panels for Sepsis Diagnosis:
1. SeptiCyte LAB
Biomarkers (Genes):Uses a 4-gene expression panel including:
**PLAC8 (Placenta-Specific 8)**.
**CEACAM4 (Carcinoembryonic Antigen Cell Adhesion Molecule 4)**.
**LY86 (Lymphocyte Antigen 86)**.
**IL1RN (Interleukin 1 Receptor Antagonist)**.
FDA-approved 4-gene panel distinguishing sepsis from non-infectious systemic inflammation.
Provides a score based on gene expression within 24 hours of ICU admission.
Purpose: Discriminates between sepsis and non-infectious systemic inflammation (e.g., SIRS).
2. Sepsis MetaScore
The Sepsis MetaScore is an 11-gene expression signature developed to differentiate sepsis from non-infectious systemic inflammation. This score calculates an 'infection z-score' based on the expression levels of these genes, aiding in the diagnosis of sepsis.
The specific genes included in the Sepsis MetaScore are:
**TGFBI**: Transforming Growth Factor Beta Induced
**SLC39A8**: Solute Carrier Family 39 Member 8
**RHAG**: Rh Associated Glycoprotein
**DDIT4**: DNA Damage Inducible Transcript 4
**FAIM3**: Fas Apoptosis Inhibitory Molecule 3
**PLAC8**: Placenta-Specific 8
**CEACAM4**: Carcinoembryonic Antigen-Related Cell Adhesion Molecule 4
**LY86**: Lymphocyte Antigen 86
**IL1RN**: Interleukin 1 Receptor Antagonist
**NLRP1**: NLR Family Pyrin Domain Containing 1
**IDNK**: Gluconokinase
These genes were selected based on their differential expression patterns in sepsis patients compared to those with non-infectious inflammation, providing a molecular basis for distinguishing between these conditions
3. FAIM3:PLAC8 Ratio
Biomarkers (Genes):
**FAIM3** (Fas Apoptosis Inhibitory Molecule 3): Linked to immune modulation.
**PLAC8** (Placenta-Specific 8): Plays a role in inflammation.
Differentiates between bacterial and viral infections.
Limited by its low negative predictive value.
4. TriVerity (InSep):
A 29-gene panel with a turnaround time of less than 30 minutes.(In article)
Identifies:Presence of infection.
Type (bacterial or viral).
Risk of mortality.
5. sNIP Panel
Biomarkers (Genes):(In article)
**NLRP1** (NLR Family Pyrin Domain Containing 1): Innate immune sensor.
**IDNK** (Gluconokinase): Related to metabolic regulation.
**PLAC8** (Placenta-Specific 8): Involved in immune response.
6. IL-8 and CCL4 Panels
Biomarkers (Proteins):
**Interleukin-8 (IL-8)**: Correlates with survival in pediatric septic shock.
**Chemokine Ligand 4 (CCL4)**: A chemokine involved in immune signaling.
7. Stratification of Patients with Sepsis
Transcriptomic-Based Subclassification:Transcriptomic data has been used to define molecular subgroups in sepsis, leading to improved patient stratification for tailored treatment.
A. Sepsis Response Signature (SRS):
The Sepsis Response Signature (SRS) is a gene expression-based framework designed to stratify patients with suspected infections into distinct molecular groups, aiding in prognosis and personalized treatment strategies. Developed by the Knight Lab, the SRS framework has been implemented in the R package SepstratifieR, which utilizes specific gene signatures to classify patients into three groups:
SRS1: Characterized by an immunosuppressed profile, often associated with a higher risk of mortality.
SRS2: Exhibits an immunocompetent profile, typically linked to a lower risk of mortality.
SRS3: Comprises mostly healthy individuals.
The SepstratifieR package offers two gene signatures for patient stratification:
7-Gene Signature (Davenport et al., 2016):
**ARL14EP** (ENSG00000152219)
**CCNB1IP1** (ENSG00000100814)
**DYRK2** (ENSG00000127334)
**ADGRE3** (ENSG00000131355)
**MDC1** (ENSG00000137337)
**TDRD9** (ENSG00000156414)
**ZAP70** (ENSG00000115085)
19-Gene Extended Signature (Cano-Gamez et al., 2021): This includes the original 7 genes plus an additional 12 genes:
**SLC25A38** (ENSG00000144659)
**DNAJA3** (ENSG00000103423)
**NAT10** (ENSG00000135372)
**THOC1** (ENSG00000079134)
**MRPS9** (ENSG00000135972)
**PGS1** (ENSG00000087157)
**UBAP1** (ENSG00000165006)
**USP5** (ENSG00000111667)
**TTC3** (ENSG00000182670)
**SH3GLB1** (ENSG00000097033)
**BMS1** (ENSG00000165733)
**FBXO31** (ENSG00000103264)
The SepstratifieR package aligns patient gene expression data to a reference dataset and employs random forest models to assign patients to one of the SRS groups. Additionally, it calculates a quantitative sepsis response score (SRSq) ranging from 0 to 1, with higher scores indicating a greater risk of adverse outcomes.
This stratification approach has been validated across various infectious diseases, including bacterial and viral sepsis, H1N1 influenza, and COVID-19, demonstrating its utility in identifying patients with dysfunctional immune profiles and facilitating precision medicine in infection management.
B. MARS (Molecular Diagnosis and Risk Stratification of Sepsis):
The Molecular Diagnosis and Risk Stratification of Sepsis (MARS) project aimed to enhance the understanding of sepsis by identifying distinct molecular endotypes among patients. Through comprehensive gene expression profiling, researchers categorized sepsis patients into four endotypes, labeled Mars1 through Mars4. Each endotype is characterized by unique gene expression patterns, which have implications for patient prognosis and potential therapeutic strategies.
Identified four endotypes (Mars1–4) using a 140-gene signature.
Mars1 patients showed the highest mortality.
MARS overlaps partially with SRS classifications but highlights population and methodological differences.
Mars1 Endotype: Gene expression ratio of BPGM (bisphosphoglycerate mutase) and TAP2 (ATP binding cassette subfamily B transporter).
Mars2 Endotype: GADD45A (growth arrest and DNA damage inducible alpha) and PCGF5 (polycomb group ring finger 5).
Mars3 Endotype: AHNAK (AHNAK nucleoprotein) and PDCD10 (programmed cell death 10).
Mars4 Endotype: IFIT5 (interferon-induced protein with tetratricopeptide repeats 5) and GLTSCR2/NOP53 (glioma tumor suppressor candidate region gene 2/ribosome biogenesis factor).
### Challenges in Sepsis Diagnostics
Timing of Sampling:
Gene expression changes dynamically over time, making it difficult to define an exact stage of disease based on transcriptomics.
The “moment in time” of sample collection may not align with disease progression.
Data Heterogeneity:
Variation in populations, sample processing, and bioinformatics analyses affects reproducibility and generalizability.
Public datasets help improve robustness but require harmonization.
Clinical Translation:
Despite promising transcriptomic data, clinical translation is slow due to:
Cost of large-scale validation studies.
Lack of calibration for clinical decision-making.
### The Genomic Storm and Heterogeneity in Sepsis
Genomic Storm:
Sepsis triggers a "genomic storm," where both pro-inflammatory and anti-inflammatory cytokines are expressed simultaneously.
This dysregulated immune response can lead to life-threatening organ dysfunction.
Heterogeneity:
Gene expression studies reveal significant variability in sepsis due to factors such as:
Host parameters (e.g., genomic variation, comorbidities).
Source of infection.
Stage of illness.
This variability has been a major challenge in clinical trials and therapeutic model development.
|
{
"filename": "251124sepsis Panels.ipynb",
"repository": "TanyaJohary/sepsis-diagnosis",
"query": "transformed_from_existing",
"size": 17292,
"sha": ""
}
|
# 01_genes.ipynb
Repository: galicae/comandos
# genes
> Prepare and annotate genes and gene sets.
<code>
# | default_exp genes
</code>
<code>
# | hide
%load_ext autoreload
%autoreload 2
</code>
<code>
# | export
import os
from typing import Union
import anndata as ad
import numpy as np
import pandas as pd
</code>
<code>
# | hide
if "EXAMPLE_DATA_PATH" not in os.environ.keys():
os.environ[
"EXAMPLE_DATA_PATH"
] = "/Users/npapadop/Documents/repos/comandos/example_data/"
</code>
SAMap is built on pairwise gene similarity. For cross-species comparisons it is often very
interesting to know the level of conservation of two genes - mostly, whether they are orthologs or
paralogs. I will provide functions to annotate genes with orthology information from EggNOG-mapper,
but you can also use your own orthology annotation, provided it returns the same thing: a $G_1
\times G_2$ table where $G_1$ and $G_2$ are the genes in the two species you are comparing, and each
cell $(g_1, g_2)$ contains the orthology relationship between $g_1$ and $g_2$: 2, if they are
orthologs, 1 if they are paralogs/in the same gene family, 0 if they are unrelated.
This is an extremely inefficient way of saving this matrix, and it would be relatively easy to code
this as a sparse matrix, but I don't think it's worth the effort. The matrices are not too large
(especially after the gene filtering that is so common to scRNA-seq analysis), and even home
computers commonly pack 16GB of RAM these days. Unless you are working with Frankenstein'd genomes
with tens of thousands of "genes" you will be fine.
First we will need to read in the EggNOG-mapper result file.
<div class="alert alert-block alert-warning">
<details>
<summary>
<b>WARNING - EggNOG format:</b>
</summary>
Depending on the version of EggNOG you may get a slightly different file; you will need to filter it
down to the two columns we need: the query gene ID and the orthogroup assignments. These are a
comma-separated string in the format `orthogroup_ID@taxonomic_level`. The taxonomic level will
determine whether genes are orthologs or paralogs, so choose wisely. I am using `"Eukaryota"` and
`"Bilateria"` as defaults, but it may well be that your version of EggNOG is using NCBI taxonomic
IDs instead of verbose names. Please check before applying!
</details>
</div>
<div class="alert alert-block alert-warning">
<details>
<summary>
<b>WARNING - Index matching:</b>
</summary>
For the entries of the EggNOG table to match to the gene names in the SAMap object we need to make
sure the index of `query` matches to the index of `sm.sams[query_species].adata.var`. If you created
the files in the scheme that I am following, this means that you prepended the species ID to the
gene IDs; we would need to do the same here.
</details>
</div>
<code>
query = pd.read_csv(
os.environ["EXAMPLE_DATA_PATH"] + "eggnog/hydra.tsv",
sep="\t",
engine="python",
)
query = query[
["Unnamed: 0", "eggNOG_OGs"]
].copy() # I am only keeping the columns I need
query.columns = ["gene_id", "eggNOG_OGs"] # rename so that it is easier to work with
query["gene_id"] = "hy_" + query["gene_id"].astype(str)
</code>
<code>
query
</code>
Now we will filter the `EggNOG_OGs` column and only keep the two levels that we're interested in.
Since we are comparing _Hydra_ to a planarian we should be using the Metazoa level for orthologs. We
can see from the table visualization that this table uses NCBI tax IDs, so we should look up the tax
ID for Metazoa (33208). Similarly, if we would like to use the Eukaryota level for paralogs, we need
its tax ID (2759).
We also need a function that will filter the orthology table to only keep the OGs that belong to the
specified levels:
<code>
# | export
def filter_OGs(
x: Union[
list, str
], # list of the EggNOG OGs or comma-separated string of EggNOG OGs.
paralog: str = "Eukaryota", # the level of the paralog OG
ortholog: str = "Bilateria", # the level of the ortholog OG
) -> list: # the paralog OG and ortholog OG
"Find the EggNOG OGs at the the paralog and ortholog level."
paralog_OG = ""
ortholog_OG = ""
if isinstance(x, str):
x = x.split(",")
for s in x:
if paralog in s:
paralog_OG = s
if ortholog in s:
ortholog_OG = s
return [paralog_OG, ortholog_OG]
</code>
This function will filter one EggNOG string (or list) to the specified levels.
<code>
query["eggNOG_OGs"].loc[0]
</code>
<code>
input_str = query["eggNOG_OGs"].loc[0]
paralog_str, ortholog_str = filter_OGs(input_str, paralog="2759", ortholog="33208")
assert paralog_str == "KOG2877@2759"
assert ortholog_str == ""
</code>
<code>
input_list = query["eggNOG_OGs"].loc[0].split(",")
paralog_list, ortholog_list = filter_OGs(input_list, paralog="2759", ortholog="33208")
assert paralog_list == "KOG2877@2759"
assert ortholog_list == ""
</code>
<code>
# | export
def assign_homology(
species_OGs, # the dataframe with the gene_id and the EggNOG OGs
paralog: str = "Eukaryota", # the level of the paralog OG
ortholog: str = "Bilateria", # the level of the ortholog OG)
) -> pd.DataFrame: # the dataframe with the gene_id, paralog OG and ortholog OG
"Get the taxonomy of the genes."
homologs = (
species_OGs["eggNOG_OGs"]
.apply(filter_OGs, paralog=paralog, ortholog=ortholog)
.to_list()
)
taxonomy = pd.DataFrame(homologs, columns=["paralog", "ortholog"])
species_OGs = species_OGs.join(taxonomy)
orthologs = pd.DataFrame(
species_OGs.groupby("gene_id", observed=False)["ortholog"].apply(np.unique).str[0]
)
paralogs = pd.DataFrame(
species_OGs.groupby("gene_id", observed=False)["paralog"].apply(np.unique).str[0]
)
result = orthologs.join(paralogs)
result.replace(to_replace="", value=None, inplace=True)
return result
</code>
<code>
hydra_genes = assign_homology(query, paralog="2759", ortholog="33208")
</code>
<code>
hydra_genes
</code>
<code>
assert hydra_genes.loc["hy_t10003aep"]["paralog"] == "KOG3599@2759"
assert hydra_genes.loc["hy_t10003aep"]["ortholog"] == "3BCY5@33208"
</code>
Repeat for the target species (planarian):
<code>
target = pd.read_csv(
os.environ["EXAMPLE_DATA_PATH"] + "eggnog/planarian.tsv",
sep="\t",
engine="python",
)
target = target[
["Unnamed: 0", "eggNOG_OGs"]
].copy() # I am only keeping the columns I need
target.columns = ["gene_id", "eggNOG_OGs"] # rename so that it is easier to work with
target["gene_id"] = "pl_" + target["gene_id"].astype(str)
planarian_genes = assign_homology(target, paralog="2759", ortholog="33208")
</code>
Given the orthology assignments (orthogroup membership), it is now very easy to calculate which
cross-species genes are orthologs or paralogs; we just need to compare the columns of the orthology
tables and keep score.
<code>
# | exporti
def compare_orthology(query, target):
result = []
for x in query:
result.append(target == x)
result = pd.DataFrame(result)
result.index = query.index
return result
</code>
<code>
# | export
def calculate_orthology_score(
query: pd.DataFrame, # the dataframe with the gene_id, paralog OG and ortholog OG for the query species
target: pd.DataFrame,
) -> pd.DataFrame:
orthologs = compare_orthology(query["ortholog"], target["ortholog"])
paralogs = compare_orthology(query["paralog"], target["paralog"])
orthology_score = orthologs.astype(int) + paralogs.astype(int)
return orthology_score
</code>
<code>
orthology_score = calculate_orthology_score(hydra_genes, planarian_genes)
</code>
<code>
gene1 = "pl_dd_Smed_v4_10002_0_1"
gene2 = "hy_t25984aep"
annot1 = target.set_index("gene_id").loc[gene1]
annot2 = query.set_index("gene_id").loc[gene2]
print(annot1["eggNOG_OGs"])
print(annot2["eggNOG_OGs"])
</code>
These genes are orthologs, we should therefore expect them to have an orthology score of 2:
<code>
assert orthology_score[gene1].loc[gene2] == 2
</code>
We can save the orthology table to disk for later use:
```python
# not run
orthology_score.to_csv("path/to/hypl_orthology.tsv", sep="\t")
```
<code>
# | export
def get_orthologs(
genes: np.ndarray, orthology: pd.DataFrame, target: ad.AnnData, celltype_to: str
) -> np.ndarray:
"""
Get orthologous and paralogous gene connections based on the given genes and orthology
information.
Parameters
----------
genes : np.ndarray
Array of gene names.
orthology : pd.DataFrame
Data frame representing the orthology information. The index should contain the query genes,
the columns should overlap with the index of target.var, and the values should be 1 for
paralogs and 2 for orthologs.
target : ad.AnnData
Target annotation data.
celltype_to : str
The target cell type. Must be a key in `target.uns["rank_genes_groups"]["names"]`.
Returns
-------
np.ndarray
Array of connections between genes, including orthologous and paralogous connections.
Columns are (query, target, degree), where degree is 1 for paralogs and 2 for orthologs.
"""
genes_in_table = np.intersect1d(genes, orthology.index)
to_include = np.sum(orthology.loc[genes_in_table]) == 2
orthologs = np.intersect1d(orthology.columns[to_include], target.var.index)
to_include = np.sum(orthology.loc[genes_in_table]) == 1
paralogs = np.intersect1d(orthology.columns[to_include], target.var.index)
scores = pd.DataFrame(target.uns["rank_genes_groups"]["scores"])[celltype_to]
names = pd.DataFrame(target.uns["rank_genes_groups"]["names"])[celltype_to]
framed = pd.DataFrame(scores).set_index(names)
significant = framed.loc[paralogs] > 0.01
homologs = np.concatenate((orthologs, paralogs[significant[celltype_to]]))
subset = orthology.loc[genes_in_table][homologs].melt(ignore_index=False)
subset.reset_index(drop=False, inplace=True)
connections = np.array(subset[subset["value"] > 0])
not_in_table = np.setdiff1d(genes, orthology.index)
if not_in_table.size > 0:
unconnected = np.array([[g, None, 0] for g in not_in_table])
return np.concatenate((connections, unconnected))
else:
return connections
def get_orthologs_overlap(genes1, genes2, query, target, orthology):
"""
Returns a DataFrame of homologous gene pairs between two sets of genes based on their presence
in an orthology table.
Parameters
----------
genes1 : numpy.ndarray
A series of gene names.
genes2 : numpy.ndarray
A series of gene names.
query : anndata.AnnData
An AnnData object containing the query genes as indices of the `.var` slot.
target : anndata.AnnData
An AnnData object containing the target genes as indices of the `.var` slot.
orthology : pandas.core.frame.DataFrame
A DataFrame containing the orthology information.
Returns
-------
connections : pandas.core.frame.DataFrame
A DataFrame of homologous gene pairs and their degree of conservation. The array has
three columns: 'query', 'target', and 'degree', where 'query' and 'target' are the gene
names, and 'degree' is the degree of conservation, which can be either 1 or 2.
"""
genes1_in_data = np.intersect1d(genes1, query.var.index)
genes2_in_data = np.intersect1d(genes2, target.var.index)
genes1_in_table = np.intersect1d(genes1_in_data, orthology.index)
genes2_in_table = np.intersect1d(genes2_in_data, orthology.columns)
connections = []
subset = orthology.loc[genes1_in_table][genes2_in_table]
_qo, _to = np.where(subset == 2)
for q, t in zip(subset.index[_qo], subset.columns[_to]):
connections.append([q, t, "2"])
_qo, _to = np.where(subset == 1)
for q, t in zip(subset.index[_qo], subset.columns[_to]):
connections.append([q, t, "1"])
return np.array(connections)
</code>
<code>
# | hide
import nbdev
nbdev.nbdev_export()
</code>
|
{
"filename": "01_genes.ipynb",
"repository": "galicae/comandos",
"query": "transformed_from_existing",
"size": 29453,
"sha": ""
}
|
# OLINK_preproc.ipynb
Repository: eduff/BIACOB
<code>
import pickle as pkl
import pandas
import pandas as pd
from pandas.api.types import is_numeric_dtype
from pandas.api.types import is_bool_dtype
import numpy as np
import pickle as pkl
import os,re
import matplotlib.pyplot as plt
import scipy.stats as stats
import sklearn
import statsmodels
import statsmodels.formula.api as smf
import seaborn as sns
import xarray
#import GPy
from scipy.stats import chi2_contingency
import numbers,numpy.random
from statsmodels.multivariate.pca import PCA
from sklearn.datasets import make_regression
import warnings
warnings.filterwarnings(action='once')
warnings.filterwarnings("ignore", category=DeprecationWarning)
warnings.simplefilter(action='ignore', category=pd.errors.PerformanceWarning)
pd.set_option('use_inf_as_na',True)
#%load_ext autoreload
#%autoreload 2
</code>
<code>
os.chdir('/data/biobank/Proteomics/OLINK')
OLINK_data=pd.read_csv('olink_data2.txt',sep='\t')
eids_OLINK=OLINK_data[OLINK_data['ins_index']==2]['eid'].unique()
eids_SIMOA=data.index.unique()
common_eids = np.union1d(eids_OLINK,data.index)
np.savetxt('subjects_OLINK.txt',common_eids,fmt='%10.0f')
OLINK=pd.DataFrame(index=np.unique(OLINK_data['eid']))
Plists={}
Plists[0]=[]
Plists[2]=[]
Plists[3]=[]
Plists['all'] = OLINK_data['protein_id'].unique()
for a in Plists['all']:
for b in [0,2,3]:
Plists[b].append('P_' + str(a) + '-' + str(b))
els=(OLINK_data['protein_id']==a)&(OLINK_data['ins_index']==b)
outsuff='0'
if b==2:
outsuff='pre'
elif b==3:
outsuff='post'
OLINK.loc[OLINK_data.loc[els,'eid'],'P_' + str(a) + '_' + outsuff] = OLINK_data.loc[els,'result'].values
OLINK_meta=pd.read_csv('ukb672393.csv')
OLINK_meta=OLINK_meta.set_index('eid')
OLINK_meta=OLINK_meta.loc[data.index,:]
</code>
<code>
OLINK_data[(OLINK_data['eid']==1024229) & (OLINK_data['ins_index']==1)]
</code>
<code>
len(OLINK_data['ins_index'].unique())
</code>
<code>
OLINK.columns=[a.replace('_3','_post') for a in OLINK.columns]
OLINK.columns=[a.replace('_2','_pre') for a in OLINK.columns]
</code>
<code>
# eids_OLINK=OLINK_data[OLINK_data['ins_index']==2]['eid'].unique()
# eids_SIMOA=data.index.unique()
# common_eids = np.union1d(eids_OLINK,data.index)
</code>
<code>
# Plists['all'] = list(OLINK_data['protein_id'].unique())
# # participants with too many missing vals
# parts_t0=(OLINK.loc[:,Plists[0]].notnull().sum(axis=1)>500)
# parts_t23=(OLINK.loc[:,Plists[2]].notnull().sum(axis=1)>500) & (OLINK.loc[:,Plists[3]].notnull().sum(axis=1)>500)
# common_eids=np.array([ a in data.index for a in OLINK.index])
# remain_eids = (~common_eids & parts_t0)
# common_eids = (common_eids & parts_t23 & parts_t0)
# common_eids_0 = (common_eids & parts_t23 )
</code>
<code>
coding = pd.read_csv('coding143.tsv',sep='\t',index_col=0)
coding.loc[:,'Protein']=np.nan
coding.loc[:,'Protein']=coding['meaning'].str.extract('(.*);').values
OLINK.columns=[a.replace('-post','_post') for a in OLINK.columns]
OLINK.columns=[a.replace('-pre','_pre') for a in OLINK.columns]
repl={}
for a in coding.index:
cols=OLINK.columns[OLINK.columns.str.contains('_'+str(a)+'-')]
for col in cols:
repl[col]=col.replace('_'+str(a)+'-','_'+coding.loc[a,'Protein']+'_')
OLINK=OLINK.rename(columns=repl)
OLINK.columns=[a.replace('_2','_pre') for a in OLINK.columns]
OLINK.columns=[a.replace('_3','_post') for a in OLINK.columns]
OLINK.to_csv('OLINK_data.csv')
</code>
<code>
cols=(OLINK.loc[:,OLINK.columns.str.contains('_pre')].notna().sum()>20)
cols=list(cols[cols].index.values)
eids_OLINK=(OLINK[cols].notna().sum(axis=1))>0
eids_OLINK=eids_OLINK[eids_OLINK].index.values
eids_SIMOA=data.index.unique()
common_eids = np.union1d(eids_OLINK,data.index)
cols = cols + [a.replace('_pre','_post') for a in cols] + [a.replace('_pre','_0') for a in cols]
OLINK.loc[subjs_OLINK,cols].to_csv('/data/biobank/Proteomics/OLINK/OLINK_data_COVID.csv')
</code>
|
{
"filename": "OLINK_preproc.ipynb",
"repository": "eduff/BIACOB",
"query": "transformed_from_existing",
"size": 10351,
"sha": ""
}
|
# Annotation_challenging_marker_gene_heatmaps.ipynb
Repository: AllonKleinLab/paper-data
# Recreate heatmaps of the Immunity paper with newly defined population (but SAME genes as in that paper)
## Import statements
<code>
import os,sys
import datetime
</code>
<code>
import scanpy as sc
sc.logging.print_versions()
sc.logging.print_memory_usage()
sc.settings.verbosity = 2
</code>
<code>
## This cell is run once to download my custom functions and import statements from github
#
#!git clone --depth=1 https://github.com/rapolaszilionis/utility_functions
#
## github doesn't seem to have an option to download a specific version of the repo from the history.
## So I download my utility functions and save the download time by appending it to the directory name.
## These utility functions to be shared together with the notebook.
#
#toappend = datetime.datetime.now().strftime('%y%m%d_%Hh%M')
#newname = "utility_functions_%s"%toappend
#print(newname)
#
#
## rename the py file with utility functions
#os.rename("utility_functions",newname)
</code>
<code>
# add the utility function folder to PATH
sys.path.append(os.path.abspath("../utility_functions_200517_09h14/"))
from rz_import_statements import *
import rz_functions as rz
import rz_utility_spring as srz
</code>
## Load scRNAseq data
<code>
# load counts
adata = sc.read_h5ad('../data/mito_total_counts_filt_raw_27563x40930_200517_10h29.h5ad')
</code>
<code>
# overwrite obs with the most recent version
adata.obs = rz.load_df('../data/obs_info_27563x32_201025_13h30.npz')
</code>
## Normalize counts
<code>
sc.pp.normalize_per_cell(adata,counts_per_cell_after=1e4)
</code>
## Get centroids (average expression over chosen labels)
<code>
thelabel = '*population'
# select only cells that have a non-nan population label.
# these exclude doublets, low quality cells, contaminating non-immune populations
cmask = ~adata.obs[thelabel].isna().values
print(cmask.sum(),len(cmask))
</code>
<code>
# get centroids
centroids = rz.centroids(thelabel,adata[cmask])
</code>
## Load gene lists for Immunity 2019 paper supploementary
<code>
# Load DGE results, this is table S2, first sheet, copied to tsvs
thepaths = glob.glob('../Zilionis_et_al_2019_supplementary_tables/table_s2_DGE_results_as_tsv/*.tsv')
framedict = {pth.split('/')[-1].split('.')[0]:pd.read_csv(pth,sep='\t',skiprows=1,index_col=0) for pth in thepaths}
</code>
<code>
framedict['fig3g'].head()
</code>
<code>
genedict = {}
for key,frame in framedict.items():
thecol = [i for i in frame.columns if i.startswith('shown')][0]
print(thecol,key)
print(frame.shape)
genedict[key] = frame.index[frame[thecol].astype(bool)]
print(len(genedict[key]))
</code>
## Plot heatmaps
<code>
figs = ['fig3g','fig4k', 'fig5g']
</code>
<code>
# pseudovalue used in Immunity paper was 50 tpm, i.e. 0.5 count per 10k.
#pseudo = 0.5
# let's use here 1 cp10k for consistency with the rest of the current study
pseudo = 1
</code>
<code>
# directory for saving results
!mkdir -p outputs/comparison_to_Immunity_heatmaps
</code>
<code>
# loop starts here:
# for saving heatmaps as separate excel sheets
fname = 'outputs/comparison_to_Immunity_heatmaps/heatmap_data'
figname = 'outputs/comparison_to_Immunity_heatmaps/Immunity_gene_heatmap'
print(fname)
# save excel tables with data
writer = pd.ExcelWriter('%s.xlsx'%fname)
for figkey in figs:
thelim = 2
# universal code
pops = framedict[figkey]['enriched_in'].unique()
# drop that "m" in from of population labels:
pops = [i[1:] for i in pops]
print(figkey,pops)
genes = genedict[figkey]
cmap = plt.cm.get_cmap('RdBu_r')
missing = [i for i in genes if i not in centroids.columns]
print(figkey,len(missing),'out of',len(genes),'genes missing')
genes = [i for i in genes if i not in missing]
heat = centroids.copy()
heat = heat.loc[pops,genes]
heat = heat + pseudo
# second normalize by second-max:
heat = heat/heat.apply(lambda x:sorted(x)[-2])
heat = np.log2(heat)
# plot heatmap
w = heat.shape[1]/100.+4
h = heat.shape[0]*0.8+1.
a,fig,gs = rz.startfig(w,h,rows=2)
hmap = a.matshow(heat,vmin=-thelim,vmax=thelim,cmap=cmap,aspect='auto')
rz.showspines(a)
a.set_xticks([])
a.set_yticks(np.arange(heat.shape[0]))
a.set_yticklabels(heat.index)
cbar = fig.colorbar(hmap)
cbar.outline.set_visible(False)
cbar.set_label('log2[(CP10K+1)/\n(CP10Kref+1)]',fontsize=5)
gs.tight_layout(fig)
# save excel
# a bit slow...
rz.color_dataframe_cells(heat,vmin=-thelim,vmax=thelim,cmap=cmap).\
to_excel(writer,'Immunity_%s'%figkey)
plt.savefig('%s_%s.pdf'%(figname,figkey),dpi=600)
plt.show()
writer.save()
</code>
## Plot signature scatterplots
Collapse the same genes as in the heatmaps into signatures and color the SPRING plot by signature expression
<code>
!mkdir -p outputs/spring_to_check_signatures
</code>
<code>
def plot_signature(
signature,
x,y,
s = 5,
pctilemax = 99.5,
vmax=None,
w = 3,
h = 3,
comment = '',
pad = 0,
ftype = 'pdf',
dpi = 1200.,
save=True,
show_cbar=False,
savedir='.',
):
"""
To write...
"""
order = np.argsort(signature)
signature = signature[order]
# get selected percentile within the selected cells only
if vmax is None:
vmax = np.percentile(signature,pctilemax)
print(vmax)
vmin = 0
if vmin==vmax:
signature = '#00ff00'
x = x[order]
y = y[order]
a,fig,gs=rz.startfig(w,h,frameon=False)
a.set_xticks([])
a.set_yticks([])
scatter = a.scatter(x,y,lw=0,s=s,c=signature,cmap=rz.custom_colormap(['#000000','#00ff00']),vmax=vmax,vmin=vmin)
if show_cbar:
cbar = plt.colorbar(scatter,orientation='horizontal')
cbar.outline.set_visible(False)
cbar.set_ticks([0,vmax])
cbar.ax.set_xticklabels(['0th','>%0.2fth'%pctilemax])
gs.tight_layout(fig,pad=pad)
if save:
plt.savefig(savedir+'/scatter_%s_%.2f_pctile.%s'%(comment,pctilemax,ftype),dpi=dpi)
plt.show()
</code>
<code>
#path to project directory
path1 = "/Users/rapolaszilionis/Google Drive/analyses/SPRING_dev/data/pittet"
project_dir = path1+'/CSF1Ri/'
plot_name = 'all_Cd45pos_cells/'
# load cell index
cellix = np.loadtxt(project_dir+plot_name+'/cell_filter.txt',dtype=int)
# load x,y coordinates
xy = pd.read_csv(project_dir+plot_name+'/coordinates.txt',header=None,index_col=0)
x = xy[1].values
y = -xy[2].values
</code>
<code>
# get dictionary of genes to plot again:
genedict2 = {}
for key in framedict.keys():
frame = framedict[key]
msk2 = frame.iloc[:,-2].astype(bool).values
gdict = {}
for pop in frame['enriched_in'].unique():
msk1 = (frame['enriched_in'] == pop).values
gdict[pop] = list(frame.index[msk1&msk2])
genedict2[key] = gdict
</code>
<code>
for figkey in figs:
for popkey in genedict2[figkey].keys():
#popkey = 'mN2'
gtp = genedict2[figkey][popkey][:20]
# gene mask
gmask = np.in1d(adata.var_names,gtp)
# select normalized counts
e = adata.X[cellix,:][:,gmask].toarray()
# decision: use z-scoring for combining.
z = scipy.stats.zscore(e,axis=0)
z = np.nan_to_num(z)
signature = z.mean(axis=1)
pctilemax = 99.95
print(popkey)
plot_signature(
signature,
x,y,
w=5,h=6,
s=3,
pctilemax=pctilemax,
comment = 'signature_%s_%s_v1'%(figkey,popkey),
show_cbar = True,
save = True,
ftype='png',
savedir='outputs/spring_to_check_signatures/')
print('\n\n')
</code>
<code>
!open outputs
</code>
|
{
"filename": "Annotation_challenging_marker_gene_heatmaps.ipynb",
"repository": "AllonKleinLab/paper-data",
"query": "transformed_from_existing",
"size": 263326,
"sha": ""
}
|
# 4_1.ipynb
Repository: IsidoraJevremovic/osnovi-astronomije
<a href="https://colab.research.google.com/github/IsidoraJevremovic/osnovi-astronomije/blob/main/4.ipynb" target="_parent"><img src="https://colab.research.google.com/assets/colab-badge.svg" alt="Open In Colab"/></a>
<code>
pip install ephem
</code>
<code>
from ephem import *
from pandas import *
import matplotlib.pyplot as plt
</code>
<code>
ime_zvezde = input("Unesite ime zvezde: ")
sirina = float(input("Unesite geografsku sirinu posmatrača: "))
duzina = float(input("Unesite geografsku duzinu posmatrača: "))
posmatrac = Observer()
posmatrac.lat = sirina
posmatrac.lon = duzina
zvezda = star(ime_zvezde)
posmatrac.date = now()
zvezda.compute(posmatrac)
if zvezda.circumpolar:
print(f"{ime_zvezde} je cirkumpolarna zvezda.")
elif zvezda.anticircumpolar:
print(f"{ime_zvezde} je anticirkumpolarna zvezda.")
else:
print(f"{ime_zvezde} izlazi i zalazi u toku dana.")
</code>
|
{
"filename": "4_1.ipynb",
"repository": "IsidoraJevremovic/osnovi-astronomije",
"query": "transformed_from_existing",
"size": 3483,
"sha": ""
}
|
# Brain_Mice_testing_functions_1.ipynb
Repository: shappiron/Aging
<code>
# If you are a developer, you may want to reload the packages on a fly.
# Jupyter has a magic for this particular purpose:
%load_ext autoreload
%autoreload 2
#data analysis libs
import numpy as np
import pandas as pd
pd.set_option('display.max_columns', 100)
from statsmodels.stats.multitest import multipletests as fdr_correction
from sklearn.feature_selection import f_classif
from scipy.stats import ttest_ind, wilcoxon, f_oneway
from matplotlib import pyplot as plt, ticker
from statannot import add_stat_annotation
from tqdm import tqdm
import seaborn as sns
sns.set_theme(font_scale=1.8)
#bioinf libs
from coolpuppy import coolpup
from plotpuppy import plotpup
import cooler
import bioframe
import cooltools
from cooltools import expected_cis, expected_trans
from cooltools.lib import plotting
import pyranges as pr
import gffpandas.gffpandas as gffpd
import gseapy
from gseapy import barplot, dotplot
#common libs
from glob import glob
import json
from os.path import basename, splitext
from importlib import reload
from natsort import natsorted, natsort_keygen
import pickle
import logging
logging.getLogger().disabled = True
#my stuff
import utils
from utils.anova import *
from utils.gene_intervals import bins_to_genes_intersection
from utils.plot_functions import *
from utils.hic_preproc import *
reload(utils.anova)
reload(utils.plot_functions)
reload(utils.hic_preproc)
</code>
<code>
#crucial work dirs
COOLER_FOLDER_PATH = '/tank/projects/kriukov_chromatin/HIC/coolers'
MERGED_COOLER_PATH = '/tank/projects/kriukov_chromatin/HIC/merged_coolers'
DOWNSAMPLED_FOLDER_PATH = '/tank/projects/kriukov_chromatin/HIC/downsampled'
MERGED_DOWNSAMPLED_FOLDER_PATH = '/tank/projects/kriukov_chromatin/HIC/merged_downsampled'
INSULATION_PATH = '/tank/projects/kriukov_chromatin/HIC/insulation'
TAD_PATH = '/tank/projects/kriukov_chromatin/HIC/tads'
LOOP_PATH = '/tank/projects/kriukov_chromatin/HIC/loops'
COMPARTMENTS_PATH = '/tank/projects/kriukov_chromatin/HIC/compartments'
GENOME_FOLDER_PATH = '/tank/projects/kriukov_chromatin/GENOME'
ATAC_FOLDER_PATH = '/tank/projects/kriukov_chromatin/ATAC'
EXPRESSION_PATH = '/tank/projects/kriukov_chromatin/RNA'
</code>
<code>
#import genes
gffpath = '%s/Mus_musculus.GRCm38.102.chr.gff3' % GENOME_FOLDER_PATH
annotation = gffpd.read_gff3(gffpath)
#annot_path = '%s/GCF_000001635.27_GRCm39_assembly_report.txt' % GENOME_FOLDER_PATH
#refseq to chrom annotation
#fasta_annot = pd.read_csv(annot_path, sep='\t', skiprows=28)
#refseq2chrom = dict(zip(fasta_annot['RefSeq-Accn'].values, fasta_annot['UCSC-style-name'].values))
gn = annotation.attributes_to_columns()
#gn.seq_id = gn.seq_id.map(refseq2chrom)
gn.seq_id = 'chr' + gn.seq_id.astype('str')
gn['length'] = gn['end'] - gn['start']
gn = gn[['seq_id', 'source', 'type', 'start', 'end', 'score', 'strand',
'ID', 'Name', 'description', 'length']]
gn['Name'] = gn['Name'].str.upper()
#types of sequences
gn_code = gn[gn['type']=='gene'] #protein codeing
gn_lnc = gn[gn['type']=='lnc_RNA'] #long non-coding
gn_trna = gn[gn['type']=='tRNA'] #tRNA
gn_rrna = gn[gn['type']=='rRNA'] #rRNA
</code>
<code>
RESOLUTION = 50_000
downsampled = True
merged = True
if downsampled:
if merged:
FILES = natsorted(glob('%s/*2000.mcool' % (MERGED_DOWNSAMPLED_FOLDER_PATH)))
CLRS = [cooler.Cooler(p + "::/resolutions/%d" % RESOLUTION) for p in FILES]
LBLS = [basename(c.filename).split('_')[0] for c in CLRS]
else:
FILES = natsorted(glob('%s/*2000.mcool' % (DOWNSAMPLED_FOLDER_PATH)))
CLRS = [cooler.Cooler(p + "::/resolutions/%d" % RESOLUTION) for p in FILES]
LBLS = ['_'.join(basename(c.filename).split('_')[:3]) for c in CLRS]
else:
if merged:
FILES = natsorted(glob('%s/*.mcool' % (MERGED_COOLER_PATH)))
CLRS = [cooler.Cooler(p + "::/resolutions/%d" % RESOLUTION) for p in FILES]
LBLS = [basename(c.filename).split('.')[0] for c in CLRS]
else:
FILES = natsorted(glob('%s/*.mcool' % (COOLER_FOLDER_PATH)))
CLRS = [cooler.Cooler(p + "::/resolutions/%d" % RESOLUTION) for p in FILES]
LBLS = ['_'.join(basename(c.filename).split('_')[:3]).split('.')[0] for c in CLRS]
[print(c, '\t', l) for c, l in zip(CLRS, LBLS)];
#compartments
if merged:
eigval_files = natsorted(glob(COMPARTMENTS_PATH + '/*_down_no_2_diag_50000*lam.txt'))
eigvec_files = natsorted(glob(COMPARTMENTS_PATH + '/*_down_no_2_diag_50000*vecs.txt'))
eigvec = {l:pd.read_csv(f, sep='\t') \
for l, f in zip(['KO', 'adl', 'old', 'yng'], eigvec_files)}
eigval = {l:pd.read_csv(f, sep='\t', index_col=1) \
for l, f in zip(['KO', 'adl', 'old', 'yng'], eigval_files)}
else:
eigval_files = natsorted(glob(COMPARTMENTS_PATH + '/*lam.txt'))
eigvec_files = natsorted(glob(COMPARTMENTS_PATH + '/*vecs.txt'))
eigvec = {l:pd.read_csv(f, sep='\t') \
for l, f in zip(['KO', 'adl', 'old', 'yng'], eigvec_files)}
eigval = {l:pd.read_csv(f, sep='\t', index_col=1) \
for l, f in zip(['KO', 'adl', 'old', 'yng'], eigval_files)}
#insulation score
if merged:
IS = pd.read_csv('%s/IS_merged_new_50000.csv' % INSULATION_PATH, index_col=0)
else:
#IS = pd.read_csv('%s/IS_25000.csv' % INSULATION_PATH, index_col=0)
IS = pd.read_csv('%s/IS_50000.csv' % INSULATION_PATH, index_col=0)
#consensus peaks
ATAC = pd.read_csv('%s/consensus_peaks.mLb.clN.boolean.annotatePeaks.txt' % ATAC_FOLDER_PATH, sep='\t')
#TADs
TADS = json.load(open(TAD_PATH + '/' + 'lavaburst_tads_arkuda.json', 'r'))
#diffexpressed genes
slopes = pd.read_csv('subdata/diffexp_result.csv', index_col=0)
pbulk_full = pd.read_csv('subdata/DE_results_GSE129788_old::young.csv', index_col=0)
pbulk_neur = pd.read_csv('subdata/DE_results_GSE129788_old::young_mNeur.csv', index_col=0)
sc_neur = pd.read_csv('subdata/scDE_results_GSE129788_old::young.csv', index_col=0)
sc_neur['gene'] = sc_neur['gene'].str.upper()
#gene sets
gene_sets = json.load(open('subdata/genesets/age_gene_set.gmt', 'r'))
#List of neuron specific genes
min_gene_length = 0
de_genes = sc_neur[(sc_neur.qval < 1e-3)]
sc_neur_filt = sc_neur.merge(gn_code[['Name', 'seq_id', 'start', 'end']], left_on='gene', right_on='Name')
#size filter
universe_neur_list = sc_neur_filt[sc_neur_filt['mean'] >= de_genes['mean'].min()].gene.tolist()
universe_neur_list = list(map(str.upper, universe_neur_list))
print('Universe size:', len(universe_neur_list))
#snp profiles
snp_profiles = pd.read_csv('subdata/snp_profiles.csv', index_col=0)
snp_merged = snp_profiles.copy()
snp_merged['adl'] = snp_profiles.filter(regex='WT').sum(1)
snp_merged['yng'] = snp_profiles.filter(regex='young').sum(1)
snp_merged['old'] = snp_profiles.filter(regex='old').sum(1)
snp_merged = snp_merged[['chrom', 'start', 'yng', 'adl', 'old']]
#view and chromsizes
mm10_arms = pd.read_csv('subdata/mm10_view.bed', sep='\t', names=['chrom', 'start', 'end', 'name'])
mm10_arms = mm10_arms.set_index("chrom").loc[CLRS[0].chromnames].reset_index()
mm10_chromsizes = CLRS[0].chromsizes
#construct scalings
scalings_data = {}
trans_data = {}
ignore_diags = 2
for cond, clr in zip(LBLS, CLRS):
# Calculate expected interactions for chromosome arms
expected = expected_cis(clr, view_df=None,
chunksize=1000000, nproc=18,
smooth=True, aggregate_smoothed=True,
smooth_sigma=0.1,
ignore_diags=ignore_diags)
trans = expected_trans(clr, view_df=None,
chunksize=1000000, nproc=18,
# full chromosomes as the view
)
scalings_data[cond] = expected
trans_data[cond] = trans
</code>
<code>
#arguments
genes = gn_code.sample(10, random_state=330)
# genes = gn_code[gn_code['Name'].isin(['ADCY5', 'HTT', 'AKT1', 'AKT2', 'CDKN1A'])]
print(genes['length'].mean())
genes = genes.sort_values('seq_id', key=natsort_keygen())
genes
</code>
<code>
def get_gene_pixels(clr, genes,
cis_exp=None,
trans_exp=None,
balance=False,
ignore_diags=2,
return_type='genes'): #may be gene list to input is better (not dataframe)
"""
Function returns submatrix of Hi-C matrix with pixels
corresponding only the genes in the given list.
"""
expected_column = 'count.avg' if balance else 'balanced.avg'
bins = clr.bins()[:].iloc[:, :3]
gb = bins_to_genes_intersection(bins, genes)
gb = bins.reset_index().merge(gb,
left_on=['chrom', 'start', 'end'],
right_on=['Chromosome', 'Start_b', 'End_b'])
idx = gb['index'].copy().tolist()
chroms = gb['chrom'].tolist()
clmat = clr.matrix(balance=balance)
mat = np.zeros((len(idx), len(idx)))
for i, (ix1, chr1) in enumerate(zip(idx, chroms)):
#for j, (ix2, chr2) in enumerate(zip(idx, chroms)):
for j, (ix2, chr2) in enumerate(zip(idx[i:], chroms[i:]), start=i):
dist = abs(i - j)
if dist < ignore_diags:
mat[i, j] = 0
else:
if chr1 == chr2:
#cis contact
if (cis_exp is not None) and (trans_exp is not None):
coef = cis_exp[(cis_exp['region1']==chr1) &
(cis_exp['dist']==dist)][expected_column].item()
else:
coef = 1
mat[i, j] = clmat[ix1, ix2] / coef
mat[j, i] = clmat[ix1, ix2] / coef
else:
#trans contact
if (cis_exp is not None) and (trans_exp is not None):
coef = trans_exp[(trans_exp['region1']==chr1) &
(trans_exp['region2']==chr2)][expected_column].item()
else:
coef = 1
mat[i, j] = clmat[ix1, ix2] / coef
mat[j, i] = clmat[ix1, ix2] / coef
if return_type=='genes':
mat = pd.DataFrame(mat, index=gb['Name'], columns=gb['Name'])
elif return_type=='intervals':
pass
return mat
</code>
<code>
genes
</code>
<code>
eps = 1#1e-6
balance = False
for clr1, clr2, pair in [[CLRS[1], CLRS[2], ('old', 'adl')],
[CLRS[3], CLRS[1], ('adl', 'yng')]]:
mat1 = get_gene_pixels(clr1, genes,
cis_exp=scalings_data[pair[1]],
trans_exp=trans_data[pair[1]],
balance=balance)
mat2 = get_gene_pixels(clr2, genes,
cis_exp=scalings_data[pair[0]],
trans_exp=trans_data[pair[0]],
balance=balance)
div = np.log2((mat2 + eps) / (mat1 + eps))
#plotting
fig, ax = plt.subplots(1,1, figsize=(7,5.5))
sns.heatmap(div, xticklabels=True, yticklabels=True, vmin=-5, vmax=5, cmap='coolwarm', ax=ax)
ax.set_title(f'log2({pair[0]}/{pair[1]}), resolution={RESOLUTION}')
plt.show()
</code>
<code>
div
</code>
|
{
"filename": "Brain_Mice_testing_functions_1.ipynb",
"repository": "shappiron/Aging",
"query": "transformed_from_existing",
"size": 220086,
"sha": ""
}
|
# project_index_1.ipynb
Repository: qwjaklj/scholarship
## Introduction
As a cloud of climate change continues getting closer every year, understanding the complex dynamics behind regional temperature disparities becomes more important. This paper sets out to unravel them by taking an area specific to the forecast between 2025 and 2074. The ultimate aim is to elaborate on the features of temperature trends and, at the end of the day, come up with mitigation ways to soften the adverse effects of the phenomenon.
Our story begins with the selection of Scenario 22, which is run by the Beijing Climate Center Climate System Model version 1.1 under the Representative Concentration Pathway 4.5. Scenario 22 was chosen from several available models, as it produced the lowest mean RMSE and, by extension, better predictive accuracy of our Random Forest model. This just cements our footing as we move ahead from the confidence in our analysis, using data from Scenario 22 to visualize the temperature trend in the future.
On plotting the projected temperatures, it was noted that between the years 2025 and 2074, there will be an increase in the temperatures that will be steady over the region. Apparently, the temperatures in the western region always rose above the temperatures in the eastern region, which led me to investigate what could be the cause of this difference in temperature. I concluded that the vegetation cover may be a major cause of the variation in the temperatures. To test this hypothesis further, we considered the spatial distribution of bare ground cover in the region. The results were indeed dramatic: the east, which was much more vegetated, was far cooler in temperature than the very open western part. Such an observation points toward the significance of vegetative cover in temperature moderation and actually warrants even greater effort to identify the actual types of vegetative covers that could be responsible for such an effect.
The next step of our research was to find out the types of vegetation that marked the most significance in regard to temperature. Having correlated the different types of vegetation with temperature, we found out that vegetative litter was the lynchpin factor. We found a significant inverse correlation of vegetative litter with fractions of summer and winter temperatures, hence implying that higher coverage of litter will result in lower temperatures. In addition, herb coverage was also found to have a significant positive correlation with litter coverage; this implies that the actions of these types of vegetation are jointly exerting an influence on the temperature levels of the study area.
Here, we provide some actionable understandings of the mechanisms by which vegetation influences temperature dynamics. Such results emphasize the importance of maintaining and increasing plant litter and herb coverage as highly effective means to fight against increasing temperatures for environmental integrity. We shall, therefore, proceed with the manner of using these insights in the development of targeted interventions in this regard that possibly may help in mitigating adverse climate change impacts and fostering a resilient ecosystem for the future.
|
{
"filename": "project_index_1.ipynb",
"repository": "qwjaklj/scholarship",
"query": "transformed_from_existing",
"size": 4163,
"sha": ""
}
|
# Career_2.ipynb
Repository: annanya-mathur/Career-Prediction
<code>
!pip install pandas
!pip install sklearn
!pip install xlrd
from sklearn.model_selection import train_test_split
from sklearn.linear_model import LinearRegression
import numpy as np
import pandas as pd
from sklearn.ensemble import RandomForestRegressor
</code>
<code>
df=pd.read_csv('exams (2).csv')
df.head(5)
</code>
<code>
career=pd.read_csv('Career_Choices.csv')
career.head(15)
</code>
<code>
df.shape
</code>
<code>
career.shape
</code>
<code>
X=df[['aptitude score','test preparation course']]
</code>
<code>
Y = df[['GradePoints']]
</code>
<code>
X_train,X_test,Y_train,Y_test =train_test_split(X,Y,test_size=0.3)
</code>
<code>
X_train
</code>
<code>
Y_test
</code>
<code>
rf=rf=RandomForestRegressor()
</code>
<code>
X_train.info()
</code>
<code>
Y_train.info()
</code>
<code>
rf.fit(np.array(X_train).reshape(-1,2),np.array(Y_train).reshape(-1,1))
</code>
<code>
a=int(input("Enter Aptitude score of student "))
b=int(input("Say whether student completed test preparation : 1 for Yes & 0 for No "))
r=np.array(rf.predict(np.array([[a,b]])))
</code>
<code>
print (r)
</code>
<code>
if r>=0.9 and r<=1.0 :
choices = career[['90% - 100%','70% - 90%','50% - 70%','40% - 50%','10% - 40%']]
elif r >=0.7 and r<=0.9:
choices = career[['70% - 90%','50% - 70%','40% - 50%','10% - 40%']]
elif r>= 0.5 and r<=0.7:
choices = career[['50% - 70%','40% - 50%','10% - 40%']]
elif r>=0.4 and r<=0.5:
choices = career[['40% - 50%','10% - 40%']]
else :
choices =career[['10% - 40%']]
final=choices.values.tolist()
</code>
<code>
type(choices)
</code>
<code>
n=len(final)
n
</code>
<code>
for i in final:
for h in i:
print(h)
</code>
<code>
import pickle
with open("career2.pkl","wb") as f:
pickle.dump(rf,f)
loader_model=pickle.load(open("career2.pkl","rb"))
result=loader_model.score(X_test,Y_test)
print(result)
</code>
<code>
h=pd.read_pickle('career2.pkl')
</code>
<code>
h
</code>
|
{
"filename": "Career_2.ipynb",
"repository": "annanya-mathur/Career-Prediction",
"query": "transformed_from_existing",
"size": 36414,
"sha": ""
}
|
# CustomDB_MTG_Taxa_Profiling_v1.0-checkpoint_1.ipynb
Repository: new-atlantis-labs/Metagenomics
# Re-formatting plankton-specific marker genes fetched from different sources to create a custom database (DB) compatible with the powerful metagenomics-based taxonomic profiling tool [Motus](https://www.nature.com/articles/s41467-019-08844-4).
See Motus' GitHub repo [here](https://github.com/motu-tool/mOTUs).
### NOTE: given that DB customization for Motus is not clearly explained in the Docs, we use a tool called [read_counter](https://github.com/AlessioMilanese/read_counter), which is a wrapper to run Motus using a customized DB.
Importantly, the default reference DB of marker genes used by Motus is not suitable for profiling marine planktonic ecosystems. Therefore we will create a plankton-specific marker gene DB to quantify relative abundance profiles across taxonomic groups. To achieve this, we will build on two well curated marker gene DBs:
- The huge catalog of phytoplankton psbO marker gene sequences, which encodes the manganese-stabilising polypeptide of the photosystem II oxygen evolving complex, reported in this [paper](https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13592) and accessible [here](https://www.ebi.ac.uk/biostudies/studies/S-BSST761?query=A%20robust%20approach%20to%20estimate%20relative%20phytoplankton%20cell%20abundances%20from%20metagenomes).
- The [MZGdb](https://metazoogene.org/MZGdb) database and most specifically the "All Plankton Combo" files contain all data from the All Zooplankton and the All Ichthyoplankton combined files. This database was described in this [paper](https://link.springer.com/article/10.1007/s00227-021-03887-y). Here we will focus on DNA sequences for the barcode region of mitochondrial cytochrome oxidase I (COI).
#### The code developed in this Notebook is meant for developing our first proof-of-concept (POC1) biodiversity data asset, which focuses on the taxonomic composition found in a given environmental sample. In a nutshell, we assess relative abundances across numerous plankton taxonomic groups from metagenomics (MTG) datasets.
</br>
Author: jay@newatlantis.io
<code>
# install for outside requirements
!pip3 install -r requirements.txt
</code>
<code>
# imports
import matplotlib
%matplotlib inline
import matplotlib.pyplot as plt
from matplotlib import cm
import seaborn as sns
import colorsys
from matplotlib.collections import PatchCollection
import Bio.SeqIO as bioseqio
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
#from Bio.Alphabet import IUPAC
from Bio import Entrez
from ete3 import NCBITaxa
from taxonomy_ranks import TaxonomyRanks
from subprocess import Popen, call, STDOUT, PIPE
import os
import shutil
import pandas as pd
import numpy as np
import matplotlib
import json
import glob
import re
import gzip
import sys
import csv
import time
import io
import pathlib
from collections import OrderedDict
import pickle
import bz2
from IPython.display import Image
from itertools import combinations
import itertools
</code>
<code>
# pandas setup
pd.set_option('mode.chained_assignment', None)
</code>
<code>
# matplotlib setup
matplotlib.rcParams['savefig.dpi'] = 1000
matplotlib.rcParams['figure.dpi'] = 1000
sns.set_style("whitegrid", {'axes.grid' : False})
sns.set_context("paper")
sns.set(font='serif')
sns.set_style('ticks')
</code>
<code>
# graphic
import plotly.graph_objects as go
import plotly.io as pio
import plotly.express as px
###Uncomment below if necessary
rendef = "png" #"pdf"
fig_renderer = pio.renderers[rendef]
fig_renderer.width = 1000
fig_renderer.height = 1000
pio.renderers.default = rendef
</code>
Utility functions
<code>
def format_tax_lbl(taxid = '94617'):
# Function to properly format taxonomic labels compatible with MetaPhlan
tax_lvls_lbls = ['species','genus','family', 'order', 'class', 'phylum', 'superkingdom'][::-1]
rank_taxon = TaxonomyRanks(taxid)
rank_taxon.get_lineage_taxids_and_taxanames()
rank_dict = list(rank_taxon.lineages.values())[0]
tax_tree = list(rank_taxon.lineages.values())[0]
#Parsing info on high rank tanks is optional, but can be quite handy when low ranks are unclassified: for higher rank assignment purposes if needed
tax_ranks_list = [tax_tree[t][0].replace("NA","unclassified") for t in tax_lvls_lbls]
ncbi_taxIDs_list = [str(tax_tree[t][1]) for t in tax_lvls_lbls]
return ncbi_taxIDs_list,tax_ranks_list
def parse_lineage(lineage_str):
# Function to correctly parse lineage using NCBI tax ID
return (";".join(format_tax_lbl(lineage_str.split(';')[-1].replace('_', ' '))[-1])).replace(" ","_")
</code>
<code>
# *Always* tell NCBI who you are
Entrez.email = "REPLACE WITH EMAIL"
</code>
<code>
# Dumping our select marker gene set into this fasta
custom_db_fasta_fid = '../custom_db/CustomPhytoZooPlanktonMGs.fna'
</code>
### Processing/filtering COI (mitochondrial cytochrome oxidase I gene) sequences for Zooplankton sepecies
<code>
coi_zooplankt_df = pd.read_csv("../data/MZGdata-coi__MZGdbALL__o00__A.csv", header=None)
</code>
A COI sequence from this highly curated gene sequence database looks like this
<code>
print(coi_zooplankt_df.iloc[1,30])
</code>
<code>
print("This COI DB contains a total number of {} sequences".format(coi_zooplankt_df.shape[0]))
</code>
The following chunks of code will perform filtering and reformatting of the headers of each sequence in the original COI DB
<code>
###Uncomment below to rebuild custom DB
filtered_coi_zooplankt_df = coi_zooplankt_df[coi_zooplankt_df.iloc[:,33].map(lambda s: isinstance(s, str))]
#Lets concentrate on organism name and corresponding COI sequence
min_coi_zooplankt_df = filtered_coi_zooplankt_df.iloc[:,[1,8,30]]
#Rename columns
#Use genebank accession to fetch a bunch of info needed to reformat headers
min_coi_zooplankt_df.columns = ["Species_name","Genebank_accession","Species_COI_seq"]
#Add full lineage
min_coi_zooplankt_df['Full_lineage'] = filtered_coi_zooplankt_df.iloc[:,33].map(lambda s: ";".join([l for l in s.split(';') if '_EXT' not in l and l!='']))
#Drop duplicate entries for genebank accessions
min_coi_zooplankt_df = min_coi_zooplankt_df.drop_duplicates(['Species_name'])
#Reset index
min_coi_zooplankt_df.reset_index(drop=True, inplace=True)
</code>
Iterating over each row in the filtered DF and creating a Seq object with formatted header, which is dumped into the dedicated file
<code>
###Uncomment below to rebuild custom DB
# for i,seq_rec in min_coi_zooplankt_df.iterrows():
# record = SeqRecord(
# Seq(seq_rec['Species_COI_seq']),
# id = seq_rec['Genebank_accession'] + '__' + seq_rec['Full_lineage'],
# name="",
# description="")
# with open(custom_db_fasta_fid, "a") as output_handle:
# bioseqio.write(record, output_handle, "fasta")
</code>
### Processing/filtering psbO sequences for Phytoplankton sepecies.
Adding to fasta file already created above
<code>
#Original psbO DB
psbO_db_fid = '../data/psbO_20210825.fna'
</code>
<code>
print("An entry in the psbO DB (fasta format) looks as follows:\n")
!head $psbO_db_fid -n 10
</code>
<code>
#Uncomment below to rebuild custom DB
with open(psbO_db_fid, "r") as handle:
for (i,record) in enumerate(bioseqio.parse(handle, "fasta")):
if(i>10):
#Fetch standard header components and reformat full header
seq_id, tax_lin = record.description.split(' ')[:2]
record.description=''
record.id = "{}__{}".format(seq_id, tax_lin)
#Dump to fasta
with open(custom_db_fasta_fid, "a") as output_handle:
bioseqio.write(record, output_handle, "fasta")
</code>
### Our final DB (concatenated gene markers for both Phyto and Zooplankton species)
<code>
print("Total number of marker genes included in our customized DB of marker genes is:")
!grep -c '>' $custom_db_fasta_fid
</code>
### With the customized marker gene DB (for both zooplankton --COI sequences-- & phytoplankton --psbO sequences--) created above we can screen across metagenomic datasets in order to assess the taxonomic composition of the plankton community sequenced.
#### A short read in a MTG file looks like this:
```@ERR1719507.4222 H2:D1NNJACXX:6:1101:5303:2333/1
AGCGAGCCCACTGTGTTCCCGGGGGACTGGGGGCCATTAGCGGCGTCAGACACGGGGGGGAGCGGGGTCTGACCATCCTGGGCCGGGACCCGGCCGTCCAGTTTGTCCAGCATGGCCCGGGCCGCCCCGTGCTTGGCCTGCTTCTTG
+
CCCFFFFFHHHHGGIIJJJJIJJJJGHIJJJJHDDDDDDDDDDCDBJJJHIGJJJJJJJJJJIJJJJJJJJIJJJJJJJJJJJJJJJEJJJJJJJJJJJJJDDDDDDDDDDDDDDDDDFFHJJIIJJJJJIJJJHGHHHFFFFFCCC```
### Analysis
The following is a simple visualiztion illustrating the relative abundances across a bunch of plankton taxonomic groups (down to the species level) that we obtained from profiling a given TARA Ocean sample (ID: [ERR1719507](https://www.ebi.ac.uk/ena/browser/view/ERR1719507)) collected in the North Atlantic Ocean (offshore Cadiz, Spain, [Location 36.5533 N 6.5669 W](https://www.google.com/maps/place/36%C2%B033'11.9%22N+6%C2%B034'00.8%22W/@36.5534368,-6.5668384,17z/data=!4m5!3m4!1s0x0:0x9aa20881883fdb5f!8m2!3d36.5533!4d-6.5669)), on date/time=2009-09-15T18:00, using a PUMP (High Volume Peristaltic Pump). The sample material (particulate matter, including plankton (ENVO:xxxxxxxx)) was collected at a depth of 38-42 m, targeting a deep chlorophyll maximum layer (ENVO:xxxxxxxx) in the marine biome (ENVO:00000447). The sample was size-fractionated (0.8-5 micrometres), and stored in liquid nitrogen for later detection of unicellular eukaryote (protist) nucleic acid sequences by pyrosequencing methods, and for later metagenomics/transcriptomics analysis. This sample has replicate sample(s): TARA_X000000407."
<code>
with open('../results/ERR1719507_mapped_reads.map','r') as fid:
lines = fid.readlines()
#Parsing lines
data = [re.split('\t|__',l.strip()) for l in lines if ';' in l]
#Pick only those with a given format to avoid noise
data = [l for l in data if len(l)==3]
</code>
<code>
#Cast data into a DF
abund_df = pd.DataFrame(data, columns = ['GeneID','Lineage','Abundance'])
#Change data type
abund_df['Abundance'] = abund_df['Abundance'].astype(float)
#Sort data by Abundance
abund_df.sort_values('Abundance', ascending=False, inplace=True)
#Reset index for tractability purpose
abund_df.reset_index(drop=True, inplace=True)
#Reformatting lineage using NCBI taxID
abund_df['Lineage'] = abund_df['Lineage'].map(parse_lineage)
#Count total number of observations/hits across unique taxa
unique_taxa_abund_df = abund_df.groupby('Lineage')['Abundance'].sum().sort_values(ascending=False)
#Take log10 and make df
unique_taxa_log_abund_df = unique_taxa_abund_df.map(np.log10).reset_index()
#Cut off by a certain value
thresholded_df = unique_taxa_log_abund_df[unique_taxa_log_abund_df['Abundance']>=1]
</code>
Make a new DF with columns = taxonomic level, and then append at the end the abundance observed in the sample analyzed
<code>
tax_enumeration_df_filtered = pd.DataFrame.from_records(thresholded_df['Lineage'].map(lambda s: s.split(';')).values)
#Name columns
tax_enumeration_df_filtered.columns = ['species','genus','family', 'order', 'class', 'phylum', 'superkingdom'][::-1]
#Add log-transformed abundance column
tax_enumeration_df_filtered['log_abundance'] = thresholded_df['Abundance'].values
</code>
<code>
#Peek at the new DF
tax_enumeration_df_filtered.head()
</code>
<code>
fig = px.sunburst(tax_enumeration_df_filtered,#.query('superkingdom == "Eukaryota"'),
path=['superkingdom','phylum', 'class', 'order', 'family', 'genus'],
values='log_abundance', color='order')
fig.update_layout(
title={
'text': "Species richness in TARA Ocean's sample (ID = ERR1719507)",
'y':0.985,
'x':0.5,
'xanchor': 'center',
'yanchor': 'top',
'font_size':30,
'font_color':"black"})
# fig.update_yaxes(automargin=True)
# fig.update_xaxes(automargin=True)
fig.update_layout(
autosize=False,
# width=500,
# height=500,
margin=dict(
l=1,
r=1,
b=4,
t=50,
pad=2
),
paper_bgcolor="White",
)
fig.show(width=1000, height=1000)
# pio.write_image(fig, "CustomProkEukDB/SunburstTaxDist_DB_v2.3.png", width=1.5*1000, height=1*1000, scale =1.25)
</code>
### Observations
- Zooplankton is clearly the numerically dominant taxonomic group in this sample.
- Based on the test performed above, one can conclude that the taxonomic profiling tools is quite effective at profiling the taxonomic composition of MTG datasets using our customized DB.
- The tool is ready for large-scale testing (using more TARA Ocean MTG datasets) for better assessing the ability of the computational pipeline to characterize taxonomic diversity across samples collected across a great variety of oceanic provinces.
- Once testing is achieved, we can confidently deploy the tool to characterize our future in-house collected MTG datasets.
|
{
"filename": "CustomDB_MTG_Taxa_Profiling_v1.0-checkpoint_1.ipynb",
"repository": "new-atlantis-labs/Metagenomics",
"query": "transformed_from_existing",
"size": 22671,
"sha": ""
}
|
# Workflows_WERONIKA_JASKOWIAK_day_1_2.ipynb
Repository: weronikajaskowiak/Comp
# Computational Workflows for biomedical data
Welcome to the course Computational Workflows for Biomedical Data. Over the next two weeks, you will learn how to leverage nf-core pipelines to analyze biomedical data and gain hands-on experience in creating your own pipelines, with a strong emphasis on Nextflow and nf-core.
Course Structure:
- Week 1: You will use a variety of nf-core pipelines to analyze a publicly available biomedical study.
- Week 2: We will shift focus to learning the basics of Nextflow, enabling you to design and implement your own computational workflows.<br>
- Final Project: The last couple of days, you will apply your knowledge to create a custom pipeline for analyzing biomedical data using Nextflow and the nf-core template.
## Basics
If you have not installed all required software, please do so now asap!
If you already installed all software, please go on and start answering the questions in this notebook. If you have any questions, don't hesitate to approach us.
1. What is nf-core?
It's a community that collects pipelines built with the nf-core language.
2. How many pipelines are there currently in nf-core?
Currently, there are 122 pipelines, but 66 are released.
3. Are there any non-bioinformatic pipelines in nf-core?
There are non-bioinformatic pipelines in nf-core, i.e. for economics and astronomy.
4. Let's go back a couple of steps. What is a pipeline and what do we use it for?
It's an automated structure consisting of modular steps. It calls existing tools to perform specific tasks. Information (input/output) is passed from one step to another according to a set of rules. We use the pipeline to automate processes and make it accessible for many people to use.
5. Why do you think nf-core adheres to strict guidelines?
The nf-core adheres to strict guidelines to maintain reproducibility, especially to obtain consistent results across different operating systems.
6. What are the main features of nf-core pipelines?
The main features are: documentation, CI Testing, stable releases, packaged software, portable and reproducible and cloud-ready.
## Let's start using the pipelines
1. Find the nf-core pipeline used to measure differential abundance of genes
<code>
# run the pipeline in a cell
# to run bash in jupyter notebooks, simply use ! before the command
# e.g.
!pwd
# For the tasks in the first week, please use the command line to run your commands and simply paste the commands you used in the respective cells!
</code>
<code>
# run the pipeline in the test profile using docker containers
# make sure to specify the version you want to use (use the latest one)
####1
!nextflow run nf-core/differentialabundance -profile test,docker --outdir /Users/weronikajaskowiak/Desktop/practical_course_2
</code>
<code>
# repeat the run. ###2
!nextflow run nf-core/differentialabundance -profile test,docker --outdir /Users/weronikajaskowiak/Desktop/test_repeated
</code>
What did change? The second runtime was faster. Moreover, the physical memory usage and CPU usage were different.
<code>
# now set -resume to the command. ###3
!nextflow run nf-core/differentialabundance -profile test,docker --outdir /Users/weronikajaskowiak/Desktop/test_repeated -resume
</code>
Check out the current directory. Next to the outdir you specified, what else has changed?
Next to the processes we have information about cached files. After opening the report, 1 file is succeeded (that one task was executed successfully during the current run and that task was not previously completed or was marked as failed in the last execution) and 20 is cached - the term "cached" indicates that the outputs from these tasks are stored and available for use without needing to reprocess them. The runtime was much shorter (around 30 sec).
<code>
# delete the work directory and run the pipeline again using -resume. What did change?
!nextflow run nf-core/differentialabundance -profile test,docker --outdir /Users/weronikajaskowiak/Desktop/test_repeated_without_work -resume
</code>
What changed? The run time was similar to the repeated one (second subtask). All files succeeded. When the work directory was deleted, nextflow cannot find any previously cached results. In this case, it treats the run as a fresh execution. All tasks will be executed anew.
## Lets look at the results
### What is differential abundance analysis?
Differential abundance analysis is a method used to determine whether the abundance (presence and quantity) of specific feature or genes - differs significantly between two or more groups or conditions.
Give the most important plots from the report:



|
{
"filename": "Workflows_WERONIKA_JASKOWIAK_day_1_2.ipynb",
"repository": "weronikajaskowiak/Comp",
"query": "transformed_from_existing",
"size": 283504,
"sha": ""
}
|
# RAG_1.ipynb
Repository: robbarto2/GenAI-Foundations
# Retrieval augmented generation (RAG)
## Loading Documents
A first step in RAG is to load document. You need a loader that supports the document type you are interested in. We use in this example Langchain, because it includes a collection of 60+ libraries for multiple types of documents and formats.
A first example with the `PyPDFLoader` library. Pdf support is direct and a single command is enough.
<code>
# For this loading Documents part, you may need these packages installed
#!pip install langchain
#!pip install -U langchain-community
</code>
<code>
import warnings # optional, disabling warnings about versions and others
warnings.filterwarnings('ignore') # optional, disabling warnings about versions and others
#!pip install pypdf
from langchain.document_loaders import PyPDFLoader
loader = PyPDFLoader("docs/War-of-the-Worlds.pdf")
book = loader.load()
</code>
<code>
# How long is the document we loaded?
len(book)
</code>
<code>
#Looking at a small extract, one page, and a few hundred characters in that page
page = book[5]
print(page.page_content[0:500])
</code>
<code>
#Which page is it, from which document?
page.metadata
</code>
A second example with a Youtube video. There is a little more work here. The yt_dlp library will need options to know what audio format to download (we won't care much about the video part). Here we use m4a, at 192 kbps. Then the ffmpeg and ffprobe programs will isolate and stream the audio part. We will then use the OpenAI whisper library to covnert the audio into text (speech-to-text).
<code>
#!pip install --upgrade --no-deps --force-reinstall yt_dlp
#! pip install pydub
#!pip install ffmpeg
#!pip install ffprobe
#!pip install --upgrade --no-deps --force-reinstall git+https://github.com/openai/whisper.git
import os
import whisper
from yt_dlp import YoutubeDL
# Step 1: Set up the download options
url = "https://www.youtube.com/watch?v=2vkJ7v0x-Fs"
save_dir = "docs/youtube/"
output_template = os.path.join(save_dir, '%(title)s.%(ext)s')
ydl_opts = {
'format': 'bestaudio/best',
'outtmpl': output_template, # Save the file to the specified directory with a title-based name
'postprocessors': [{
'key': 'FFmpegExtractAudio',
'preferredcodec': 'm4a', # You can change this to mp3 if you prefer
'preferredquality': '192',
}],
'ffmpeg_location': '/opt/homebrew/bin/ffmpeg', # Specify the location of ffmpeg
}
# Step 2: Download the audio from the YouTube video
with YoutubeDL(ydl_opts) as ydl:
ydl.download([url])
# Step 3: Find the downloaded file
downloaded_file = [f for f in os.listdir(save_dir) if f.endswith('.m4a')][0] # Assuming m4a, adjust if using mp3
downloaded_file_path = os.path.join(save_dir, downloaded_file)
# Step 4: Load the Whisper model
model = whisper.load_model("base") # You can choose 'tiny', 'base', 'small', 'medium', or 'large'
# Step 5: Transcribe the audio file
result = model.transcribe(downloaded_file_path)
</code>
<code>
# Adding metadata to the transcript, and saving the transcript to a file so we can use it outside of this program.
class Document:
def __init__(self, source, text, metadata=None):
self.source = source
self.page_content = text
self.metadata = metadata or {}
# Wrap the transcription result in the Document class with metadata
document = Document(
source=downloaded_file_path,
text=result['text'],
metadata={"source": "youtube", "file_path": downloaded_file_path}
)
#Save the transcript to a text file
transcript_file_path = os.path.join(save_dir, 'transcript.txt')
with open(transcript_file_path, 'w') as f:
f.write(result['text'])
print(f"Transcript saved to {transcript_file_path}")
</code>
<code>
# how many characters in this transcript file?
len(document.page_content)
</code>
<code>
# Print the first 500 characters of the transcript
print(document.page_content[:500])
</code>
## Splitting our documents in chunks
A second step is to split our documents (a 128-page book and 32K-character trasncript file) into smaller chunks. We use Langchain libraries here again.
<code>
# We will use the most important library, recursive character splitter
from langchain.text_splitter import RecursiveCharacterTextSplitter
</code>
<code>
# Chunks have a character length, and an overlap values. For example (in real life, you are probably closer to 500 to 1000 and 50 to 100 respectively):
rsplit = RecursiveCharacterTextSplitter(
chunk_size=20,
chunk_overlap=5,
separators=["\n\n", "\n", "(?<=\. )", " ", ""]
)
</code>
<code>
# Let's take an example string
text1 = 'abcdefghijklmnopqrstuvwxyz1234567890'
</code>
<code>
rsplit.split_text(text1)
</code>
<code>
Hamlet = """Truly to speak, and with no addition, \
We go to gain a little patch of ground \
That hath in it no profit but the name. \
To pay five ducats, five, I would not farm it; \
Nor will it yield to Norway or the Pole \
A ranker rate, should it be sold in fee."""
</code>
<code>
rsplit.split_text(Hamlet)
</code>
<code>
# Let's go for a more realistic chunk size
rsplit = RecursiveCharacterTextSplitter(
chunk_size=500,
chunk_overlap=100,
separators=["\n\n", "\n", "(?<=\. )", " ", ""]
)
</code>
<code>
# Looking at the files, first the pdf
rdoc1 = rsplit.split_documents(book)
</code>
<code>
len(rdoc1)
</code>
<code>
# the splitted version has more documents (pages) than the original pdf source,
len(book)
</code>
<code>
#Printing a few splits
for i, doc in enumerate(rdoc1[30:33]): # Adjust the number 3 to print more or fewer splits
print(f"--- Split {i + 1} ---")
print(doc.page_content)
print() # Print an empty line for better readability
</code>
<code>
# Splitting the trasncript of the audio file
from langchain.text_splitter import RecursiveCharacterTextSplitter
from langchain.schema import Document
# Step 1: Load the transcript text
transcript_file_path = "docs/youtube/transcript.txt"
with open(transcript_file_path, 'r') as f:
transcript_text = f.read()
# Step 2: Create a Document object
document = Document(page_content=transcript_text)
# Step 3: Split the transcript into chunks
rdoc2 = rsplit.split_documents([document])
# Step 4 manually assigning the metadata to each split
save_dir = "docs/youtube/"
downloaded_file = [f for f in os.listdir(save_dir) if f.endswith('.m4a')][0] # Assuming m4a, adjust if using mp3
downloaded_file_path = os.path.join(save_dir, downloaded_file)
for doc in rdoc2:
doc.metadata = {"source": "youtube", "file_path": downloaded_file_path}
# Step 5: Print the first few splits
for i, doc in enumerate(rdoc2[30:33]): # Adjust the number 3 to print more or fewer splits
print(f"--- Split {i + 1} ---")
print(doc.page_content)
print() # Print an empty line for better readability
</code>
<code>
# Checking the metadata
# Viewing metadata of the first few splits from rdoc1 (the pdf text)
print("Metadata for rdoc1:")
for i, doc in enumerate(rdoc1[:3]): # Adjust the number to view more or fewer splits
print(f"--- Metadata for Split {i + 1} ---")
print(doc.metadata) # Print the metadata
print() # Print an empty line for better readability
# Viewing metadata of the first few splits from rdoc2 (the video transcript)
print("Metadata for rdoc2:")
for i, doc in enumerate(rdoc2[:3]): # Adjust the number to view more or fewer splits
print(f"--- Metadata for Split {i + 1} ---")
print(doc.metadata) # Print the metadata
print() # Print an empty line for better readability
</code>
Recursive character splitting is a very common technique. But if you use an LLM that severly limits the number of input token (or charges you b y the token), you may want to split based on tokens instead of character sequences. This is how to do it.
<code>
from langchain.text_splitter import TokenTextSplitter
</code>
<code>
# Let's define a very small chunk and no overlap, so you can see what a chunk looks like with this method
token_split = TokenTextSplitter(chunk_size=1, chunk_overlap=0)
</code>
<code>
print(token_split.split_text(Hamlet))
</code>
## Storing in Vector Store
The third step is to store your splits in a vector database. There are dozens of solutions. Very popular solutions for local storage include Mongodb, Chroma, Weaviate and Milvus. All large Cloud vendors (Azure, AWS etc.) offer a Cloud vectordb solution. Here we use Chroma, a locally stored, flexible popular choice.
Before storing our data into the vectordb, we need to convert the text strings into vectors (embedding). We use a tokenizer compatible with the BERT model to first tokenize the text, then embed (convert to vectors).
<code>
# Create Ollama embeddings and vector store
#!pip install chromadb
from langchain_community.vectorstores import Chroma
from langchain_community.embeddings import OllamaEmbeddings
all_splits = rdoc1 + rdoc2
embeddings = OllamaEmbeddings(model="nomic-embed-text")
vectorstore = Chroma.from_documents(documents=all_splits, embedding=embeddings)
</code>
What do these vectors look like? Let's play with a few examples.
<code>
text1 = "i like hotdogs"
text2 = "i like sandwiches"
text3 = "this is a large building"
</code>
<code>
embedding1 = embeddings.embed_query(text1)
</code>
<code>
embedding1 = embeddings.embed_query(text1)
embedding2 = embeddings.embed_query(text2)
embedding3 = embeddings.embed_query(text3)
</code>
<code>
# looking at the first values of the first embedding
print("embedding1 includes", len(embedding1), "values")
print("First few values:", embedding1[:10])
</code>
How closes are these vectors from one another? There are many ways to compare them, here we use the cosine similarity method.
<code>
import numpy as np
from numpy import dot
from numpy.linalg import norm
# Step 1 : creating the normalized vectors (so the product is between 0 and 1)
norm_a = np.linalg.norm(embedding1)
norm_b = np.linalg.norm(embedding2)
norm_c = np.linalg.norm(embedding3)
normalized_a = embedding1 / norm_a
normalized_b = embedding2 / norm_b
normalized_c = embedding3 / norm_c
#Step 2: comparing text1 and text 2 embeddings, then text1 and text 3 embeddings:
def cosine_similarity(a, b):
return dot(a, b) / (norm(a) * norm(b))
similarity_1_2 = cosine_similarity(embedding1, embedding2)
similarity_1_3 = cosine_similarity(embedding1, embedding3)
print("Similarity (with cos similarity) between sentence 1 and 2:", similarity_1_2)
print("Similarity (with cos similarity) between sentence 1 and 3:", similarity_1_3)
</code>
Now that we have embeddings, let's store them into a Chroma database.
<code>
#!pip install --upgrade langchain chromadb
from langchain.vectorstores import Chroma
# Set the environment variable to disable tokenizers parallelism and avoid warnings
os.environ["TOKENIZERS_PARALLELISM"] = "false"
# Let's define a directory where we'll store the database beyond this notebook execution (and let's make sure it is emtpy, as I run this notebook often :))
persist_directory = 'docs/chroma/'
!rm -rf ./docs/chroma # remove old database files if any
</code>
<code>
vectordb = Chroma.from_documents(
documents=all_splits,
embedding=embeddings,
persist_directory=persist_directory
)
</code>
Now let's see if we can perform some similarity search with this database. keep in mind that we are just comparing vectors here, there is no LLM yet to smartly correlate deeper.
<code>
question = "Did the spaceship come from the planet Mars?"
</code>
<code>
docs = vectordb.similarity_search(question,k=5)
</code>
<code>
len(docs)
</code>
<code>
docs[0].page_content
</code>
<code>
# Let's save the vectordb so we can use it outside of this notebook - note, this is FYI as it is automatically done with Chroma, but not with all other vectordbs!
vectordb.persist()
</code>
## Retrieving with the LLM in action
The full process consists of asking a question, retrieving the relevant information, then passing the information and the question to the LLM.
<code>
#We still need these bricks, so do not run this part of the notebook in isolation
persist_directory = 'docs/chroma/'
embedding = embeddings
vectordb = Chroma(persist_directory=persist_directory, embedding_function=embedding)
</code>
<code>
print(vectordb._collection.count())
</code>
<code>
question = "Did the spaceship come from the planet Mars?"
docs = vectordb.similarity_search(question,k=3)
len(docs)
</code>
<code>
#!pip install ollama
#!ollama serve & ollama pull llama3 & ollama pull nomic-embed-text
</code>
<code>
#Using Llama3 as the LLM, and Ollama as the wrapper to interact with Llama3. Then using a test question to calidate the install.
from langchain_community.llms import Ollama
llm = Ollama(model = "llama3")
llm.invoke("Are there aliens on Mars?")
</code>
<code>
#!pip install ollama langchain beautifulsoup4 chromadb gradio -q
</code>
<code>
# This is "almost" the final code. You will see the final code in the last lesson of the course
import gradio as gr
import ollama
from bs4 import BeautifulSoup as bs
from langchain_community.embeddings import OllamaEmbeddings
# Create Ollama embeddings and vector store
#embeddings = OllamaEmbeddings(model="nomic-embed-text")
#vectorstore = Chroma.from_documents(documents=splits, embedding=embeddings)
# Define the function to call the Ollama Llama3 model
def ollama_llm(question, context):
formatted_prompt = f"Question: {question}\n\nContext: {context}"
response = ollama.chat(model='llama3', messages=[{'role': 'user', 'content': formatted_prompt}])
return response['message']['content']
# Define the RAG setup
retriever = vectordb.as_retriever()
def rag_chain(question):
retrieved_docs = retriever.invoke(question)
formatted_context = "\n\n".join(doc.page_content for doc in retrieved_docs)
return ollama_llm(question, formatted_context)
# Define the Gradio interface
def get_important_facts(question):
return rag_chain(question)
# Create a Gradio app interface
iface = gr.Interface(
fn=get_important_facts,
inputs=gr.Textbox(lines=2, placeholder="Enter your question here..."),
outputs="text",
title="RAG with Llama3",
description="Ask questions about the provided context",
)
# Launch the Gradio app
iface.launch()
# example q: did the aliens eventually go on to land on Venus?
</code>
|
{
"filename": "RAG_1.ipynb",
"repository": "robbarto2/GenAI-Foundations",
"query": "transformed_from_existing",
"size": 43733,
"sha": ""
}
|
# 6a_Run_ChromVAR_new_snATAC_2.ipynb
Repository: Gaulton-Lab/non-diabetic-islet-multiomics
#### Summary:
This is a notebook to run chromvar on peaks derived from scATAC-seq stored in a Seurat object. This can be reworked to use peaks not entered into a Seurat object. ChromVAR needs 3 inputs. 1) A count matrix that is barcode x peak or sample x peak 2) GRanges identifying peaks and 3) motifs. Motifs can be a custom set, or downloaded from JASPAR. I have code in another location to pull and save JASPAR motifs into a usable format. JASPAR seems to update yearly. The motifs I pulled are all human.
From here there are follow-up notebooks to compare cell types and disease states.
A reference for ChromVAR can be found here: https://greenleaflab.github.io/chromVAR/articles/Introduction.html
**NOTE:** this notebook illustrates how to make inputs and run ChromVAR with Wang et al. snATAC data, we followed the same commands to do this for the HPAP snATAC data as well.
<code>
suppressMessages(library(chromVAR))
suppressMessages(library(motifmatchr))
suppressMessages(library(SummarizedExperiment))
library(Signac)
library(Seurat)
library(JASPAR2020)
library(TFBSTools)
library(BSgenome.Hsapiens.UCSC.hg38)
library(patchwork)
library(readr)
library(stringr)
library(dplyr)
set.seed(1234)
library(ggplot2)
library(Matrix)
library(BiocParallel)
register(MulticoreParam(8))
#### Send to channel code
library(parallel)
library(qvalue)
</code>
<code>
outdir <- '/dir/with/phenotype/assoc/outputs/ATAC/ChromVAR'
</code>
# 1. Create long format matrices for Wang et al. snATAC data
Did this with scripts, see notion note
# 2. Read in LFMs and make merged barcode x union peak matrix
<code>
motif.ix <- readRDS('/path/to/240322_WE_MotifMatches_Union_Peaks.RDS')
dim(motif.ix)
head(rownames(motif.ix))
</code>
## Wang snATAC
<code>
lfm_dir <- file.path(outdir,'long_format_matrices/')
list.files(lfm_dir)
</code>
<code>
#read in all LFMs and add any missing information
lfms <- list()
hvws <- rownames(motif.ix)
samples <- list.files(lfm_dir)
print(Sys.time())
for (samp in samples) {
print(paste0(which(samples %in% samp) * 100 /length(samples),"% - ", samp))
#Read in the atac lfm matrix and make sure is union peaks, also add sample prefix to barcodes
lfm_in <- read.table(paste0(lfm_dir, samp,'/',samp,'.long_fmt_mtx.txt.gz'), col.names=c('Window','Barcode','Count'))
lfm_in$Window <- as.factor(lfm_in$Window)
lfm_in$Barcode <- paste(samp,lfm_in$Barcode,sep='_')
lfm_in$Barcode <- as.factor(lfm_in$Barcode)
lfm_in_filt <- filter(lfm_in, Window %in% hvws)
print(dim(lfm_in_filt))
mis_windows <- hvws[!hvws %in% levels(lfm_in_filt$Window)]
print(length(mis_windows))
#make sure there are missing_windows
if (length(mis_windows) > 0){
print('Adding missing windows')
#create a new long format matrix (sm) with the missing windows added as 0 counts
filler_bc <- as.character(lfm_in_filt$Barcode[[1]])
print(paste("Using filler BC:",filler_bc,sep=" "))
new_rows <- cbind(as.data.frame(mis_windows),
as.data.frame(rep(filler_bc),length(mis_windows)),
as.data.frame(rep(0,length(mis_windows))))
colnames(new_rows) <- c("Window","Barcode","Count")
lfm_in_fix <- rbind(lfm_in_filt,new_rows)
#if there aren't, set lfm to df
} else {
print('No windows were missing')
lfm_in_fix <- lfm_in_filt
}
print(dim(lfm_in_fix))
#finalize matrix
lfm_in_fix$Window <- factor(lfm_in_fix$Window, levels=hvws)
lfm_in_sort <- lfm_in_fix[order(lfm_in_fix$Window),]
lfm_in_sort$Barcode <- as.factor(lfm_in_sort$Barcode)
lfms[[samp]] <- with(lfm_in_sort, sparseMatrix(i=as.numeric(Window), j=as.numeric(Barcode), x=Count, dimnames=list(levels(Window), levels(Barcode))))
print(dim(lfms[[samp]]))
gc()
print('')
}
gc()
summary(lfms)
print(Sys.time())
</code>
<code>
#make final lfm
lfm <- do.call(cbind, lfms)
dim(lfm)
head(lfm)
</code>
<code>
#how many peaks have counts -- almost all
length(names(which(rowSums(lfm) != 0)))
names(which(rowSums(lfm) == 0))
</code>
# 3. Make inputs and run ChromVAR
<code>
#Prep inputs for SummarizedExperiment
sc.data <- lfm
# Extract peak locations and reformat into GRanges object
bed = str_split_fixed(rownames(sc.data), "\\-", 3) #This may need to be modified depending on your peak naming convention
gr= GRanges(seqnames = bed[,1], ranges = IRanges(start = as.numeric(bed[,2]), end = as.numeric(bed[,3])))
</code>
<code>
# annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
# seqlevelsStyle(annotation) <- 'UCSC'
</code>
<code>
#Use the matrix and GRanges to make a SummarizedExperiment to put into chromvar and add metadata
fragment.counts <- SummarizedExperiment(assays=list(counts=sc.data), rowRanges=gr)
# fragment.counts.stored <- fragment.counts
fragment.counts <- addGCBias(fragment.counts, genome=BSgenome.Hsapiens.UCSC.hg38)
fragment.counts
</code>
<code>
#remove peaks or motifs with no counts
fragment.counts.sub <- fragment.counts[names(which(rowSums(lfm) != 0)),]
fragment.counts.sub
motif.ix.sub <- motif.ix[names(which(rowSums(lfm) != 0)),]
motif.ix.sub
</code>
<code>
# Run chromVAR -- approx 30 mins
print(Sys.time())
dev <- computeDeviations(object=fragment.counts.sub, annotations=motif.ix.sub)#, expectation=expected)#
head(dev)
print(Sys.time())
</code>
<code>
saveRDS(dev, file.path(outdir,'outputs',"250116_HM_ChromVAR_Object_Union_Peaks_Wang.RDS"))
</code>
# 4. Collect details for downstream analysis
<code>
# Load in motifs object
jaspar.motifs <- readRDS(file ='/dir/with/motif/object/jaspar_2022_object.Rdata')
</code>
<code>
dev
jaspar.motifs
</code>
<code>
#This is a cell by motif deviation score (aka accessibility) matrix
devtab = deviationScores(dev)
head(devtab)
</code>
<code>
# Variation of accessibiility across deviation scores, basically the standard errror. Null is about 1
variability <- computeVariability(dev)
head(variability)
</code>
<code>
motifdata = cbind(sapply(jaspar.motifs, function(x) unlist(x@name)),sapply(jaspar.motifs, function(x) unlist(x@matrixClass )))
motifdata # sapply(pfm, function(x) x@tags$symbol ) , sapply(pfm, function(x) x@tags$family ))
</code>
<code>
#Save devscores, motifdata, and info
write.table(devtab, file=file.path(outdir,'outputs','devscores_Union_Peaks_Wang.txt'),quote = FALSE, col.names = TRUE, row.names = TRUE)
write.table(motifdata, file=file.path(outdir,'outputs','motifdata_Union_Peaks_Wang.txt'),quote = FALSE, col.names = FALSE,sep='\t')
write.table(variability, file=file.path(outdir,'outputs','variability_Union_Peaks_Wang.txt'),quote = FALSE, col.names = TRUE, row.names = TRUE, sep='\t')
</code>
<code>
# Variability distirbution
ggplot(variability, aes(x=variability)) +
geom_histogram() + geom_vline(xintercept = 1.5)
</code>
<code>
sessionInfo()
</code>
|
{
"filename": "6a_Run_ChromVAR_new_snATAC_2.ipynb",
"repository": "Gaulton-Lab/non-diabetic-islet-multiomics",
"query": "transformed_from_existing",
"size": 80757,
"sha": ""
}
|
# stat.ipynb
Repository: ElinaZhang0721/DeepTarget-NLP
## Data Prepare and Clean
<code>
import pandas as pd
import re
# Load the XLSX file
file_path = 'C:/Users/yufei/Programming/DeepTarget/web_scrape/scraped_data.xlsx'
data = pd.read_excel(file_path)
data.head()
</code>
<code>
from bs4 import BeautifulSoup
# Remove any rows with null values in the 'Cleaned Main Content' column
data = data.dropna(subset=['Main Content'])
# Define a function to clean the 'Main Content' column
def clean_text(text):
# Remove HTML tags if present
text = BeautifulSoup(text, "html.parser").get_text()
# Remove extra spaces, tabs, and newlines
text = re.sub(r'\s+', ' ', text).strip()
return text
# Clean the 'Main Content' column
data['Cleaned Main Content'] = data['Main Content'].apply(clean_text)
data.head()
</code>
<code>
# Export the cleaned data to an Excel file
# file_path = "C:/Users/yufei/Downloads/cleaned_data.xlsx"
# data.to_excel(file_path, index=False)
</code>
<code>
filtered_data = data[['Title','URL','Cleaned Main Content']]
</code>
## Word Count
<code>
# Define a function to count the number of words in each summary
def word_count(text):
return len(str(text).split())
# Apply the word count function to the "Summary Content" column
filtered_data['Word Count'] = filtered_data['Cleaned Main Content'].apply(word_count)
filtered_data.head()
</code>
## Count the number of sentences
<code>
# Define a function to count the number of sentences in each summary
def sentence_count(text):
return len(str(text).split('. '))
# Apply the sentence count function to the "Summary Content" column
filtered_data['Sentence Count'] = filtered_data['Cleaned Main Content'].apply(sentence_count)
filtered_data.head()
</code>
## Count the number of nounces
<code>
import nltk
nltk.download('punkt')
nltk.download('averaged_perceptron_tagger')
from nltk import pos_tag
from nltk.tokenize import word_tokenize
# Tokenize and count nouns
def noun_count(text):
words = word_tokenize(str(text))
pos_tags = pos_tag(words)
nouns = [word for word, pos in pos_tags if pos in ['NN', 'NNS', 'NNP', 'NNPS']]
return len(nouns)
filtered_data['Noun Count'] = filtered_data['Cleaned Main Content'].apply(noun_count)
filtered_data.head()
</code>
## Sentiment Analysis
<code>
from textblob import TextBlob
# Define a function to get the sentiment polarity
def get_sentiment(text):
blob = TextBlob(text)
return blob.sentiment.polarity
# Apply the sentiment function to the "Summary Content" column
filtered_data['Sentiment'] = filtered_data['Cleaned Main Content'].apply(get_sentiment)
# Show the dataframe with sentiment scores
filtered_data.head()
</code>
- -1 indicates Very Negative
- -0.5 indicates Negative
- 0 indicates Neutral
- 0.5 indicates Positive
- 1 indicates Very Positive
## Readability Score - Flesch-Kincaid readability score
<code>
import textstat
# Define a function to calculate the Flesch-Kincaid readability score
def calculate_readability(text):
return textstat.flesch_reading_ease(text)
# Apply the readability function to the "Summary Content" column
filtered_data['Readability'] = filtered_data['Cleaned Main Content'].apply(calculate_readability)
# Display the first few rows with the readability scores
filtered_data.head()
</code>
- 90-100: Very easy to read
- 60-90: Easily understood by 13-15-year-olds
- 30-60: College-level text
- 30-: Very difficult to read
## Aggregated Statistics
<code>
# Count the Mean
aggregation_results = filtered_data[['Word Count', 'Sentence Count', 'Noun Count', 'Sentiment', 'Readability']].agg(
['mean']
)
# Display the aggregated results
aggregation_results
</code>
<code>
# Round the results
aggregation_results['Word Count'] = aggregation_results['Word Count'].round(0)
aggregation_results['Sentence Count'] = aggregation_results['Sentence Count'].round(0)
aggregation_results['Noun Count'] = aggregation_results['Noun Count'].round(0)
aggregation_results['Sentiment'] = aggregation_results['Sentiment'].round(4)
aggregation_results['Readability'] = aggregation_results['Readability'].round(4)
aggregation_results.head()
</code>
|
{
"filename": "stat.ipynb",
"repository": "ElinaZhang0721/DeepTarget-NLP",
"query": "transformed_from_existing",
"size": 50596,
"sha": ""
}
|
# Job_Analysis_Doc2Vec_Analysis.ipynb
Repository: jgroth1/NLP
<code>
import json
import os
from bs4 import BeautifulSoup
import re
from nltk.stem import WordNetLemmatizer
from nltk import sent_tokenize, wordpunct_tokenize, pos_tag
from nltk.corpus import stopwords
import nltk
from gensim.models.doc2vec import TaggedDocument, Doc2Vec
from gensim import corpora, models, similarities
from gensim.models.phrases import Phraser, Phrases
</code>
<code>
def paras(html):
soup = BeautifulSoup(html, 'html.parser')
for string in soup.stripped_strings:
yield string
def sents(html):
for paragraph in paras(html):
for sentence in sent_tokenize(paragraph):
yield sentence
def words(html):
for sentence in sents(html):
for token in wordpunct_tokenize(sentence):
yield token
def tokenize(html):
for paragraph in paras(html):
yield [pos_tag(wordpunct_tokenize(sent)) for sent in sent_tokenize(paragraph)]
</code>
<code>
docs = []
titles = []
stop_words = set(stopwords.words('english'))
wnl = WordNetLemmatizer()
n=0
for i in range(10):
i += 1
path = 'corpus3/02-09-2018/Page_' + str(i) + '/'
files = os.listdir(path)
rxs = [re.search('^\.',file) for file in files]
i =[n for n in range(len(rxs)) if rxs[n]]
m = 0
for k in i:
files.pop(k-m)
m += 1
for file in files:
with open(path + file, encoding='utf-8') as f:
job_post = f.read()
job_dict = eval(job_post)
job_dict.keys()
title = job_dict['job title']
company = job_dict['company']
titles.append([n, title, company])
html = job_dict['job description']
doc = []
xs = words(html)
for x in xs:
if x not in stop_words:
doc.append(wnl.lemmatize(x).lower())
docs.append(doc)
n += 1
</code>
<code>
corpus = [d for d in docs]
</code>
<code>
documents = [TaggedDocument(doc, [i]) for i, doc in enumerate(docs)]
model = Doc2Vec(documents, vector_size=50, min_count=3)
</code>
<code>
stop_words = set(stopwords.words('english'))
wnl = WordNetLemmatizer()
n=0
resses = []
with open('/Users/grothjd/documents/DS_resume/Time_Series_Resume/Jonathan_Groth_PhD_Resume_test.txt', encoding='utf-8') as f:
resume = f.read()
#print(resume)
xs = words(resume)
for x in xs:
if x not in stop_words:
resses.append(wnl.lemmatize(x).lower())
</code>
<code>
inferred_vector = model.infer_vector(resses)
sims = model.docvecs.most_similar([inferred_vector])
select = []
for sim in sims:
select.append(sim[0])
</code>
<code>
job = []
for title in titles:
if int(title[0]) in select:
job.append(title)
print(job)
</code>
<code>
keywords = model.wv.similar_by_word("data", 100)
</code>
<code>
keywords
</code>
|
{
"filename": "Job_Analysis_Doc2Vec_Analysis.ipynb",
"repository": "jgroth1/NLP",
"query": "transformed_from_existing",
"size": 12990,
"sha": ""
}
|
# C.elegans_demo_1.ipynb
Repository: kunwang34/PhyloVelo
# Run PhyloVelo in C.elegans data
We next sought to benchmark PhyloVelo by applying to the phylogeny-resolved scRNA-seq data of C. elegans. The embryonic lineage tree of C. elegans is entirely known. Moreover, time-course single-cell RNA-seq data from C. elegans embryos has been mapped to its invariant lineage tree. We focused on the AB lineage which had the densest cell annotations from generation 5 (32-cell stage) to 12 (threefold stage of development), with mostly ectoderm and accounting for ~70% of the terminal cells in the embryo.
Data are available from Packer _et al_. at the Gene Expression Omnibus (GEO) under accession number GSE126954. Since multiple synchronous C. elegans embryos were pooled for the scRNA-seq experiment, many nodes on the embryonic lineage tree have been sampled multiple times. Only one cell was thus randomly chosen to represent the corresponding node, while these non-repetitive cells from one lineage tree constitute a “pseudo-embryo”. We focused on the AB lineage (the largest lineage amongst the first five founder cells contributing over 70% somatic tissues of C. elegans) and filtered out the genes with total count < 10 (remaining 6533 genes and 298 cells in a single psedo-emberyo). For the dimensionality reduction embedding of the scRNA-seq data, we used the UMAP embedding coordinates from original study13, which is available from https://github.com/qinzhu/Celegans_code/blob/master/globalumap2d_Qin.rds.
<code>
import pandas as pd
import matplotlib.pyplot as plt
import phylovelo as pv
import numpy as np
from scipy.stats import spearmanr
from mpl_toolkits.axes_grid1.inset_locator import inset_axes
</code>
## import data
<code>
import pyreadr
globalumap = pyreadr.read_r('./datasets/Celegans/globalumap2d_Qin.rds')
globalumap = globalumap[None]
</code>
<code>
cell_annotation = pd.read_csv('./datasets/Celegans/GSE126954_cell_annotation.csv', index_col=0)
count = pd.read_csv('./datasets/Celegans/pseudoembryo0_count.csv', index_col=0)
sample = count.index
</code>
## Read data using scData and filter
<code>
sd = pv.scData(count=count, Xdr=globalumap.loc[sample], cell_generation=cell_annotation.loc[sample].lineage.apply(lambda x: len(x.split('/')[0])).to_numpy().flatten())
sd.drop_duplicate_genes(target='count')
sd.normalize_filter(is_normalize=False, is_log=False, min_count=10, target_sum=None)
</code>
## Inference and project velocity into embedding
<code>
pv.velocity_inference(sd, sd.cell_generation, cutoff=0.95, target='count')
pv.velocity_embedding(sd, target='count', n_neigh=15)
</code>
<code>
fig, ax = plt.subplots()
scatter = ax.scatter(sd.Xdr.iloc[:, 0], sd.Xdr.iloc[:, 1], c=sd.cell_generation, s=100)
ax = pv.velocity_plot(sd.Xdr.to_numpy(), sd.velocity_embeded, ax, 'stream',streamdensity=1.2, grid_density=25, radius=0.12, lw_coef=10000)
ax.figure.set_size_inches(8,8)
ax.set_xlabel('UMAP 1', fontsize=15)
ax.set_ylabel('UMAP 2', fontsize=15)
cbaxes = inset_axes(ax, width="3%", height="30%", loc='lower right')
plt.colorbar(scatter, cax=cbaxes, orientation='vertical')
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
ax.set_ylim([0.3, 1.65])
</code>
## Inference PhyloVelo pseudotime
<code>
sd = pv.calc_phylo_pseudotime(sd, n_neighbors=280)
</code>
<code>
fig, ax = plt.subplots()
ax.figure.set_size_inches(8,8)
scatter = ax.scatter(sd.Xdr.iloc[:,0], sd.Xdr.iloc[:,1], s=100, c=sd.phylo_pseudotime,cmap='plasma')
ax.set_xlabel('UMAP 1', fontsize=15)
ax.set_ylabel('UMAP 2', fontsize=15)
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
cbaxes = inset_axes(ax, width="3%", height="30%", loc='lower right')
plt.colorbar(scatter, cax=cbaxes, orientation='vertical')
</code>
<code>
x = cell_annotation.loc[sample]['embryo.time'].to_numpy()
y = sd.phylo_pseudotime
a, b = np.polyfit(x, y, deg=1)
y_est = a * np.linspace(50,650,60) + b
fig, ax = plt.subplots(figsize=(7, 6))
ax.scatter(x, y, alpha=0.4, s=70)
ax.plot(np.linspace(50,650,60), y_est, '-', c='k')
ax.set_xlabel('Embryo time', fontsize=17)
ax.set_ylabel('PhyloVelo pseudotime', fontsize=17)
# ax.text(50, 0.9, 'spearmanr={:.2g}\np_val={:.2g}'.format(*spearmanr(x, y)), fontsize=15)
rho, pval = spearmanr(x, y)
ax.text(50, 0.98, r"Spearman's $\rho$={:.2g}".format(rho), fontsize=15)
ax.text(50, 0.91, r'$p={}\times 10^{}$'.format(*r'{:.2g}'.format(pval).split('e')).replace('^', '^{').replace('$', '}$')[1:], fontsize=15)
ax.spines['right'].set_visible(False)
ax.spines['top'].set_visible(False)
</code>
|
{
"filename": "C.elegans_demo_1.ipynb",
"repository": "kunwang34/PhyloVelo",
"query": "transformed_from_existing",
"size": 247265,
"sha": ""
}
|
# x.ipynb
Repository: ArhamNaeem/Timetable-Project
<code>
import pandas as pd
</code>
<code>
teachers = pd.read_csv('teachers.csv')
rooms= pd.read_csv('room.csv')
classes = pd.read_csv('class.csv')
</code>
<code>
teachers
</code>
<code>
rooms
</code>
<code>
classes
</code>
<code>
timetable = {}
</code>
|
{
"filename": "x.ipynb",
"repository": "ArhamNaeem/Timetable-Project",
"query": "transformed_from_existing",
"size": 10524,
"sha": ""
}
|
# analysis_Ratz2022Clonal_2.ipynb
Repository: yuanzhiyuan/SODB
Clonal relations in the mouse brain revealed by single-cell and spatial transcriptomics
ShortName: Ratz2022Clonal
Steps of processing the data from raw to Anndata:
<code>
# 1, Download the raw data from GSE153424
</code>
<code>
# 2, Unzip the .tar file to get:
# xxx_filtered_feature_bc_matrix.h5
# xxx_raw_feature_bc_matrix.h5
# xxx_scalefactors_json.json.gz
# xxx_tissue_hires_image.png.gz
# xxx_tissue_positions_list.csv.gz
# ...
</code>
<code>
# 3, For each experiment, organize the path structure as:
# - Experiment Name
# - filtered_feature_bc_matrix.h5
# -spatial
# - scalefactors_json.json
# - tissue_hires_image.png
# - tissue_positions_list.csv
</code>
<code>
# 4, Run SCANPY code to load and write Anndata
import scanpy as sc
adata = sc.read_visium(raw_path)
adata.write_h5ad(h5ad_path)
</code>
<code>
# The data is now transform from raw to Anndata, then it should be processed to be accepted to SODB
</code>
<code>
# Run ShortName.ipynb in Anndata2SODB path
</code>
|
{
"filename": "analysis_Ratz2022Clonal_2.ipynb",
"repository": "yuanzhiyuan/SODB",
"query": "transformed_from_existing",
"size": 2832,
"sha": ""
}
|
# in_BioMedical_Ask_BioRxiv_1.ipynb
Repository: compu-flair/LLMs
<code>
# installs
#pip install langchain tiktoken openai langchainhub chromadb
</code>
<code>
import requests
import json
from bs4 import BeautifulSoup
import os
</code>
<code>
## change the
query = "single cell RNA sequencing"
print(type(query))
start_date = "2010-01-01" ## "YYYY-MM-DD"
end_date = "2023-12-30"
# Replace with the actual bioRxiv API endpoint for searching articles
url = f"https://api.biorxiv.org/details/biorxiv/{start_date}/{end_date}"
# Parameters for the API request
params = {
'q': query, # your search query
'num_results': 100 # number of results to return
}
# Send a request to the bioRxiv API
response = requests.get(url, params=params)
</code>
<code>
# Check if the request was successful
if response.status_code == 200:
# Parse the response JSON
number_of_articles = 100 # Filter the first 10 articles
data = response.json()
# Extract article information
articles = data['collection'] # Adjust this based on actual response structure
articles = articles[:number_of_articles]
# Create a directory for articles if it doesn't exist
if not os.path.exists('articles'):
os.makedirs('articles')
for article in articles:
#print(f"Title: {article['title']}")
if article['published'] == "NA":
continue
print(f"Title: {article['title']}")
doi = article['doi']
# Replace / with _ in DOI to make it a valid filename
filename = doi.replace('/', '_') + '.txt'
# Include the folder name in the path
filepath = os.path.join('articles', filename)
# bioRxiv metadata
full_text_url = f"https://www.biorxiv.org/content/{doi}.full"
try:
response = requests.get(full_text_url)
if response.status_code == 200:
soup = BeautifulSoup(response.content, 'html.parser')
texts = soup.get_text(separator=' ', strip=True)
# Writing the texts to a file inside the articles folder
with open(filepath, 'w', encoding='utf-8') as file:
file.write(texts)
print(f"Text written to {filepath}")
except Exception as e:
print(f"Problem reading the article: {e}")
</code>
<code>
from langchain.vectorstores import Chroma
from langchain.embeddings import OpenAIEmbeddings
from langchain.text_splitter import RecursiveCharacterTextSplitter
from langchain.llms import OpenAI
from langchain.chains import RetrievalQA
from langchain.document_loaders import TextLoader
from langchain.document_loaders import DirectoryLoader
</code>
<code>
#Load from a directory
loader = DirectoryLoader('./articles/', glob="./*.txt", loader_cls=TextLoader) #- We will use this in case of many articles saved in a directory
documents = loader.load()
</code>
<code>
#splitting the text into smaller documents
text_splitter = RecursiveCharacterTextSplitter(chunk_size=1000, chunk_overlap=0)
texts = text_splitter.split_documents(documents)
</code>
<code>
# Lets see how many smaller documents we have created from the large document
len(texts)
</code>
<code>
# Lets select the 4th document to see how it looks as a small chunk
texts[50]
</code>
<code>
import os
from dotenv import load_dotenv
import chromadb
#from langchain_community.embeddings.sentence_transformer import SentenceTransformerEmbeddings # use openai embeddings instead
#from langchain_community.vectorstores import Chroma # We will use this as our local vectorstore
# Load environment variables from a .env file
load_dotenv()
OPENAI_API_KEY = os.getenv('openai_api_key')
# Embed and store the texts
## here we are using OpenAI embeddings but we could also use the sentensetransformer embeddings
embedding_function = OpenAIEmbeddings(model = 'text-embedding-ada-002',openai_api_key=OPENAI_API_KEY)
# create the open-source embedding function
#embedding_function = SentenceTransformerEmbeddings(model_name="all-MiniLM-L6-v2")
# load it into Chroma
db = Chroma.from_documents(texts, embedding_function)
</code>
<code>
# Making a retriever
def retriever(query, num_chunks):
retriever = db.as_retriever(search_kwargs={"k": num_chunks})
docs = retriever.get_relevant_documents(query) # Here we are filtering documents with similar meaning to the query
return docs
</code>
<code>
def print_sources(documents):
seen_dois = set() # A set to keep track of DOIs that have already been printed
for doc in documents:
# Extract the 'source' from the document's metadata
source = doc.metadata['source']
# Extract the DOI part from the 'source'
parts = source.split('/')[1].split('.') # Split into parts by '.' after 'articles/'
doi = '.'.join(parts[:-1]) # Join all parts except the last one ('txt')
# Replace underscore with slash in DOI
doi = doi.replace('_', '/')
# Print the DOI if it hasn't been seen before
if doi not in seen_dois:
print( f'Relevant Docs: Doi: {doi} ')
seen_dois.add(doi) # Add the DOI to the set of seen DOIs
</code>
<code>
# Set your OpenAI API key here
OPENAI_API_KEY = os.getenv('openai_api_key')
from openai import OpenAI
client = OpenAI(
api_key=OPENAI_API_KEY
)
</code>
<code>
def answer_question(content, question):
# Construct the conversation
messages = [{"role": "system",
"content": """
You are a helpful research assistant.
You will be given a Content and a Question. Answer the Question by rephrasing and smoothening the Content.
Do not add any information but what is in the documents.
If your output do not meaningfully answer the question, say irrelevant documents.
"""
},
{"role": "user", "content": content},
{"role": "user", "content": question}
]
# Make the API call
try:
response = client.chat.completions.create(
model="gpt-3.5-turbo-1106", #"gpt-4-1106-preview"
messages=messages,
temperature=1 # creativity
)
return response.choices[0].message.content
except Exception as e:
print(f"An error occurred: {e}")
return None
</code>
<code>
def get_content(query):
docs = retriever(query, num_chunks=5)
content = "Content: "
for doc in docs:
content += doc.page_content if hasattr(doc, 'page_content') else ''
return content
</code>
<code>
# Asking a new question
subject = "enzymatic pre-amplification"
question = f"Question: what do you know about {subject}?"
content = get_content(query)
answer_question(content, question)
</code>
<code>
# Asking a new question
subject = "airplane"
question = f"Question: what do you know about {subject}?"
content = get_content(query)
answer_question(content, question)
</code>
|
{
"filename": "in_BioMedical_Ask_BioRxiv_1.ipynb",
"repository": "compu-flair/LLMs",
"query": "transformed_from_existing",
"size": 15844,
"sha": ""
}
|
# Submodule08_Differential_Analysis_Proteomics_2.ipynb
Repository: NIGMS/Analysis-of-Biomedical-Data-for-Biomarker-Discovery
<img src="images/RIINBRE-Logo.jpg" width="400" height="400"><img src="images/MIC_Logo.png" width="600" height="600">
# Analysis of Biomedical Data for Biomarker Discovery
<a id="top8"></a>
## Submodule 8: Identification of IRI Biomarkers from Proteomic Data
### Dr. Christopher L. Hemme
### Director, [RI-INBRE Molecular Informatics Core](https://web.uri.edu/riinbre/mic/)
### The University of Rhode Island College of Pharmacy
---
## Overview
This Jupyter Notebook performs a differential expression analysis on proteomic data to identify biomarkers for Ischemia-Reperfusion Injury (IRI). Using the `limma` package in BioConductor, the notebook explores the impact of surgical treatment and a drug treatment (treprostinil) on protein expression levels over time. It begins with data preparation, loading a pre-processed experimental object containing normalized proteomic data and metadata. A linear regression model is built, first considering treatment alone, then incorporating time as a combined covariate. The analysis utilizes MA plots, volcano plots, and the `topTable` function to visualize and rank differentially expressed proteins. The notebook concludes by discussing the next steps in biomarker discovery, including pathway analysis, meta-analysis, experimental validation, and comparison to known biomarkers, highlighting the increasing role of machine learning in handling the growing complexity of biomedical data.
## Learning Objectives
1. **Data Preparation for Differential Analysis:** How to prepare proteomic data and metadata for analysis in R, including loading necessary packages, reading data from saved objects, combining covariates, and cleaning data.
2. **Linear Regression for Differential Expression:** How to use the `limma` package in Bioconductor to perform linear regression-based differential expression analysis, accounting for batch effects using `duplicateCorrelation` and `lmFit`.
3. **Working with Contrasts:** How to define and use contrasts to extract pairwise comparisons from the global regression model, enabling more accurate and sophisticated comparisons between different experimental conditions.
4. **Interpreting MA Plots and Volcano Plots:** How to generate and interpret MA plots and volcano plots to visualize differential expression results, understand data distribution, assess normalization effects, and identify significantly changed proteins.
5. **Extracting Top Differentially Expressed Proteins:** How to use `topTable` to extract the most significantly differentially expressed proteins based on log2 fold change and adjusted p-values.
6. **Considering Time and Treatment Effects:** How to combine time and treatment covariates to analyze the dynamic changes in protein expression due to treatment over time.
7. **Next Steps in Biomarker Discovery:** Understanding the broader context of biomarker discovery beyond differential expression analysis, including pathway analysis, meta-analysis, and experimental validation.
8. **Introduction to Machine Learning for Biomarker Discovery:** Briefly introduces the emerging role of machine learning in biomarker discovery and sets the stage for the next submodule.
## Prerequisites
1. **Create a Vertex AI Notebooks instance:** Choose your machine type and other configurations.
2. **Select the R kernel:** Make sure you choose the appropriate R environment when creating or opening the notebook.
3. **Install R Packages:** The notebook itself handles the installation of the necessary R packages (`devtools`,`statmod`,`preprocessCore`,`limma`,`ComplexHeatmap`,`M3C`,`EnhancedVolcano`,`Glimma`,`plyr`,`dplyr`,`matrixStats`,`tidyverse`)
4. **Data Storage:** Upload your data to a Cloud Storage bucket and ensure your service account has access to read the data.
## Introduction
In this submodule we will complete our analysis of the IRI proteomics data by caring out a linear regression based differential analysis (also called differential expression analysis) using the <b>limma</b> package in BioConductor. In differential analysis, we are looking for significant changes in specific features between one or more states. We measure these changes by comparing the "peaks" of each feature across the states. A peak represents whatever signal we're measuring. For proteomics and metabolomics, this is usually the signal intensity from a mass spectrometry machine. For next-generation sequencing experiments, these peaks will be based on the read counts from your alignment step. A peak can represent a variety of signals depending on your experiment, but what matters is that the peak represents some biological quantity.
In comparing two peaks for the same feature across two states, we are interested in whether the difference in peak heights are statistically significant (measured by the <i>p</i>-value) and the magnitude of this difference. This second value is a measure of the <b>effect size</b> and in omics studies typically the log2(fold change) between the two samples. We can then use <i>p</i>-value and log2(fold change) cutoffs to identify which proteins significantly differ between states. While this may sound complicated, to get these values we use the same linear regression methods we studied in Chapter 3 and the coefficients of our regression model are the log2(fold change) values.
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> Blue boxes will indicate helpful tips.</div>
<div class="alert alert-block alert-warning">
<b>🎓 Fun Facts:</b> Used for interesting asides or notes.
</div>
<div class="alert alert-block alert-success">
<b>👊 Success:</b> This alert box indicates a successful or positive action.
</div>
<div class="alert alert-block alert-danger">
<b>🛑 Caution:</b> A red box indicates potential hazards or pitfalls you may encounter.
</div>
---
## Get Started
### Data Prep
We will start with our usual data prep (installation of packages, loading of experimental object, definition of variables).
<code>
if(!require(devtools)) install.packages("devtools")
devtools::install_github("cran/statmod")
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
library('BiocManager')
BiocManager::install("preprocessCore")
BiocManager::install("preprocessCore", configure.args="--disable-threading", force = TRUE)
bioc_packages <- c("limma", "ComplexHeatmap", "M3C", "EnhancedVolcano", "Glimma")
installed_bioc_packages <- bioc_packages %in% rownames(installed.packages())
if (any(installed_bioc_packages == FALSE)) {BiocManager::install(bioc_packages[!installed_bioc_packages])}
packages <- c("plyr", "dplyr", "matrixStats", "statmod")
installed_packages <- packages %in% rownames(installed.packages())
if (any(installed_packages == FALSE)) {install.packages(packages[!installed_packages])}
</code>
<code>
require('preprocessCore')
require('limma')
require('Glimma')
require('ComplexHeatmap')
require("EnhancedVolcano")
require('M3C')
require('tidyverse')
require('plyr')
require('matrixStats')
require('statmod')
</code>
<code>
exp_obj <- readRDS(file = "data/Saved_Data/exp_obj.rds")
</code>
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> The proteome and metadata objects loaded below are saved in the .rds object in previous submodules. If you see blank variables for either, make sure you've run through the code in submodule 6 and 7 to have the most up to date saved variables in <i>exp_obj.rds</i></div>
<code>
proteome <- exp_obj$data$proteomics$norm
metadata <- exp_obj$metadata
metadata
</code>
<code>
#Preview proteome data
head(proteome)
</code>
<div class="alert alert-block alert-danger">
<b>🛑 Caution:</b> Reminder that this version of the experimental object should contain the quantile normalized data from the last chapter. Use <i>str(exp_obj)</i> if you need to verify that this is the case.
</div>
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> Reminder:
<ul>
<li><b>CTRL</b> - Control samples, no surgery or treatment
<li><b>SHAM</b> - Surgical treatment only; Designed to see if the stress of cutting the rat causes any changes
<li><b>PLB</b> - IRI model with placebo
<li><b>TRE</b> - IRI model treated with trepostrinil
</ul>
</div>
For the sake of simplicity and because this experiment wasn't explicitly designed as a longitudinal study, we will combine our Treatment and Time covariates into a single covariate with 16 factors using a custom R function that we will call <i>group_metadata</i>. This will allow us to run a simple linear regression model using both covariates. However, you are welcome to test more complicated models such as a multivariate model with both Treatment and Time as separate covariates.
<code>
# custom function that accepts a metadata data frame and covariates to group together into a single covariate.
# Returns a new version of the metadata data frame with the new covariate column called Combined added.
group_metadata <- function(metadata, groups) {
metadata <- metadata %>%
group_by_at(groups) %>%
unite(Combined, all_of(groups), sep = "", remove = FALSE)
metadata$Combined <- factor(metadata$Combined)
metadata
}
metadata <- group_metadata(metadata, c("Treatment", "Time"))
metadata
</code>
Before we run the combined covariates model, let's start with just Treatment. We will use the cell means version of the formula which sets the intercept to 0. Because our data is log2 transformed, our regression coefficients will represent the log2 fold change of the given protein between groups. First we need to build a model matrix which takes our model and converts the categorical variables to dummy variables (0 or 1). Our model matrix will be as follows:
<code>
molecule.model.treatment <- model.matrix(~ 0 + Treatment, metadata)
colnames(molecule.model.treatment) <- levels(metadata$Treatment)
molecule.model.treatment
</code>
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> The model matrix is also called a design matrix.</div>
The next step will be to define the parameters of our regression model. We will account for the batch effect observed in the last chapter by setting the <i>Batch</i> column as a blocking factor in the function <i>duplicateCorrelation</i> which computes the intrablock correlation. Once these parameters are set, we will run a simple fit which <b>limma</b> will return as an <b>MArrayLM</b> object. Because we need to account for our blocking factor, we will first run the <i>duplicateCorrelation</i> function to estimate the the correlations between the observations of our blocking variable (i.e. our batch effect). We will then pass these results to the <i>lmFit</i> function.
<code>
molecule.corfit.treatment <- duplicateCorrelation(
proteome,
molecule.model.treatment,
block = metadata$Batch
)
</code>
<code>
molecule.SimpleFit.treatment <- lmFit(
proteome,
molecule.model.treatment,
block = metadata$Batch,
correlation = molecule.corfit.treatment$consensus
)
molecule.SimpleFit.treatment
</code>
We'll look at these results in more detail later. Right now, we need to think about <b>contrasts</b>. Typically when we want to compare two states, we would use a simple <b>t-test</b>. However, using multiple t-tests can introduce error if it's not corrected. A better strategy is to extract pairwise information from our global regression model using <b>contrasts</b>. With contrasts, we can define very sophisticated comparisons by combining different factors together, but we'll keep it simple for now and just compare different treatment states.
<code>
# define contrasts
contrasts.treatment <- c("SHAM-CTRL", "PLB-SHAM", "TRE-PLB")
# make the contrasts
molecule.contrasts.treatment <- makeContrasts(contrasts = contrasts.treatment, levels = molecule.model.treatment)
# refit the simple fit we previously ran using the contrast data
molecule.contrastsFit.treatment <- contrasts.fit(molecule.SimpleFit.treatment, molecule.contrasts.treatment)
# calculate stats and rank proteins using an emperical Bayes method
molecule.contrastsFit.treatment <- eBayes(molecule.contrastsFit.treatment)
# fix column names to match contrasts
colnames(molecule.contrastsFit.treatment$coefficients) = contrasts.treatment
</code>
The full regression model gives us a global overview of which covariates and features are important. With contrasts, we can break that information down and see the changes for each protein within the context of each specific pair of Treatment states we're interested in.
### Data Cleaning
Before continuing Lets see if there are any NA (Not Available) values in `molecule.contrastsFit.treatment$coefficients`.
<code>
nrow(molecule.contrastsFit.treatment$coefficients)
</code>
It includes `2897` rows. Lets look at the first five rows of it:
<code>
molecule.contrastsFit.treatment$coefficients[1:5,]
</code>
Now, we are going to count number of rows in the table such that it includes "at least" one NA value:
<code>
num_NA_rows <- sum(rowSums(is.na(molecule.contrastsFit.treatment$coefficients)) > 0)
num_NA_rows
</code>
We can also see which row includes a null value:
<code>
which(is.na(molecule.contrastsFit.treatment$coefficients[,1]))
</code>
Now use the output to see what column contains the NA value:
<code>
molecule.contrastsFit.treatment$coefficients[1434,]
</code>
The above analysis shows that there is one NA value in `SHAM-CTRL` column of our data.
We can safely remove the 1434th row.
<code>
molecule.contrastsFit.treatment <- molecule.contrastsFit.treatment[-1434,]
</code>
### Diagnostic plots
Before we analyze the specific protein changes, let's look at some diagnostic plots. We'll start with the <b>MA plot</b>. The MA plot shows the relationship between the log2(fold change) (the "M" in MA) and the mean expression (the "A"). We expect most proteins will not show differential expression in a given experiment, and thus most of the data points will be located at 0. Extreme values along the y-axis indicate proteins with strongly differential values between the two states. Particularly when working with genomic data, the MA plot will often show a "fanning" effect due to the fact that low expression genes (plotted on the left) show more variability in fold change than highly expressed genes (plotted on the right). MA plots are also useful for evaluating the effects of normalization on the data. Let's look at the MA plots for our three contrasts to get a sense of the landscape of our data. We'll use the <b>Glimma</b> package which is used to create interactive plots from <b>limma</b> data. You will be able to clock on specific data points to see the effects on specific genes.
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> Glimma plots are interactive. If you click on datapoints on the graph, the names of the points will appear in the box below and filter the table.</div>
<code>
#SHAM-CTRL
glimmaMA(molecule.contrastsFit.treatment, coef=1)
</code>
<code>
#PLB-SHAM
glimmaMA(molecule.contrastsFit.treatment, coef=2)
</code>
<code>
#TRE-PLB
glimmaMA(molecule.contrastsFit.treatment, coef=3)
</code>
What do these results show us? As hoped, there aren't many significant difference between the expression profiles of control and sham, meaning the surgical technique itself introduces a minimal amount of noise. As expected, the IRI itself induces many proteomic changes compared to the sham. Based on treatment alone though, there are few significant changes in protein levels for the treated state. However, we still have to consider the time component. In early time points, the changes may not be significant enough to see an effect from treatment, and in later time points the damage could be significant enough that treatment is ineffective. To get a better sense of what is happening, we will need to add the time component. First though, let's verify this with the volcano plots.
A <b>volcano plot</b> is another extremely useful visualization tool for comparing pairwise omics data. In this case, we are plotting the log2(fold change) on the x-axis and the -log10(<i>p</i>-value) on the y-axis. This centers the data on the x-axis on zero, meaning that the plot on each side of zero represents one of our factors of interest. By applying cutoff, we can easily identify quadrants of significantly different proteins and easily identify which treatment state they represent.
<code>
#SHAM-CTRL
glimmaVolcano(molecule.contrastsFit.treatment, coef=1)
</code>
<code>
#PLB-SHAM
glimmaVolcano(molecule.contrastsFit.treatment, coef=2)
</code>
<code>
#TRE-PLB
glimmaVolcano(molecule.contrastsFit.treatment, coef=3)
</code>
The results are consistent but easier to visualize on the volcano plot. It's important to remember that the volcano plot is showing <b>relative</b> abundance, that is, how abundant is a signal in one condition versus another. It doesn't tell you anything about the expression levels themselves. The MA plot and box plots (and the raw data of course) are useful for determining why this abundance occurs (e.g. a protein is more highly expressed in condition B vs. condition A).
Now we will use the <i>topTable</i> function to look at the regression coefficients themselves for SHAM-CTRL, PLB_SHAM, and finally TRE-PLB.
<code>
topTable(molecule.contrastsFit.treatment, coef = "SHAM-CTRL", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<code>
topTable(molecule.contrastsFit.treatment, coef = "PLB-SHAM", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<code>
topTable(molecule.contrastsFit.treatment, coef = "TRE-PLB", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> The "M" in the <b>sort.by</b> argument refers to the log2(fold change), or the "M" axis in our MA plot.</div>
The cumulative results show that there is a clear change in state between the sham and the placebo (i.e. untreated IRI) states, but we will need to include the time component to better understand what is happening. Let's run these analyses again, except this time we will use our combined covariate to account for time. We'll only look at the 6 hour comparisons but feel free to try different contrasts. The following code is the same as above except for the formula used to define our model. Above, we used this formula to define our model as <i>model.matrix(\~0 + Treatment, metadata)</i> whereas this below code defines the model as <i>model.matrix(\~0 + Treatment, metadata).</i>
<code>
molecule.model.combined <- model.matrix(~ 0 + Combined, metadata)
colnames(molecule.model.combined) <- levels(metadata$Combined)
molecule.model.combined
</code>
<code>
molecule.corfit.combined <- duplicateCorrelation(
proteome,
molecule.model.combined,
block = metadata$Batch
)
</code>
<code>
molecule.SimpleFit.combined <- lmFit(
proteome,
molecule.model.combined,
block = metadata$Batch,
correlation = molecule.corfit.combined$consensus
)
molecule.SimpleFit.combined
</code>
<code>
contrasts.combined <- c("SHAM6-CTRL0", "PLB6-SHAM6", "TRE6-PLB6")
molecule.contrasts.combined <- makeContrasts(contrasts = contrasts.combined, levels = molecule.model.combined)
molecule.contrastsFit.combined <- contrasts.fit(molecule.SimpleFit.combined, molecule.contrasts.combined)
molecule.contrastsFit.combined <- eBayes(molecule.contrastsFit.combined)
colnames(molecule.contrastsFit.combined$coefficients) = contrasts.combined
</code>
### Data Cleaning
Before continuing Lets see if there are any NA (Not Available) values in `molecule.contrastsFit.combined$coefficients`.
<code>
molecule.contrastsFit.combined$coefficients[1:5,]
</code>
<code>
nrow(molecule.contrastsFit.combined$coefficients)
</code>
`molecule.contrastsFit.combined$coefficients` includes 2897 rows. Lets see how many of these rows includes at least one NA (Not Available) value.
<code>
rows_with_NA=rowSums(is.na(molecule.contrastsFit.combined$coefficients)) > 0 # It is a vector of length 2897 with Boolean values
num_NA_rows <- sum(rows_with_NA)
num_NA_rows
</code>
Finally we remove rows with NA value:
<code>
molecule.contrastsFit.combined <- molecule.contrastsFit.combined[!rows_with_NA, ]
</code>
### Diagnostic plots
<code>
#SHAM6-CTRL0
glimmaMA(molecule.contrastsFit.combined, coef=1)
</code>
<code>
#PLB6-SHAM6
glimmaMA(molecule.contrastsFit.combined, coef=2)
</code>
<code>
#TRE6-PLB6
glimmaMA(molecule.contrastsFit.combined, coef=3)
</code>
<code>
#SHAM6-CTRL0
glimmaVolcano(molecule.contrastsFit.combined, coef=1)
</code>
<code>
#PLB6-SHAM6
glimmaVolcano(molecule.contrastsFit.combined, coef=2)
</code>
<code>
#TRE6-PLB6
glimmaVolcano(molecule.contrastsFit.combined, coef=3)
</code>
<code>
topTable(molecule.contrastsFit.combined, coef = "SHAM6-CTRL0", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<code>
topTable(molecule.contrastsFit.combined, coef = "PLB6-SHAM6", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<code>
topTable(molecule.contrastsFit.combined, coef = "TRE6-PLB6", number = 10, adjust.method = "BH", sort.by = "M")
</code>
<div class="alert alert-block alert-info">
<b>✋ Tip:</b> As an exercise, redo the previous analysis using the multivariate model "~ 0 + Treatment + Time" to see if separating the covariates has an affect on the results.</div>
Now we have a clearer view of the changes that are happening between the states. At 6 hours when we expect IRI damage to be noticeable but still treatable, we start to see significant differences in the protein abundances between the sham and placebo states, but we also see several proteins affected by the trepostrinil treatment. While we can try different models to identify other covariates affected the results, we have now identified a set of potential biomarkers that can indicate the degree of severity of IRI and other biomarkers which may indicate the effects of the treatment on IRI damage.
---
# Finishing Our Experiment
You should now be familiar with the concepts of linear and logistic regression and how they can be used to compare biomarkers or to classify samples or to assess the quality of biomarkers. We have discussed common methods of exploratory analysis such as PCA and heatmaps that are useful for identifying global patterns in omics data. Finally, we discussed differential analysis of omics data to identify specific potential biomarkers. However, we are ending on something of a cliffhanger. Now that we have a list of potential protein biomarkers from our proteomics analysis, what comes next?
First, we need to assess the biological relevance of our results. A common way to do this is with <b>pathway analysis</b>. Most proteins are part of complex metabolic and regulatory networks that represent some biological theme (e.g. TCA cycle, apoptosis, etc.). We would expect proteins within a specific pathway or biological process to be correlated together, otherwise it could lead to significant disruption of the cellular machinery. Just as we can test for differential abundance of proteins, we can also look at differential abundance of pathways. In our case, several of the proteins we find deferentially expressed are related to kidney injury and function, which is what we would expect. With this biological knowledge, we can begin to filter our list of potential biomarkers to include thode that are most likely to be biological relevant in our model (i.e. causative of or directly caused by the state change).
Ideally, we would design our experiments to have sufficient numbers of samples and controls to maximize the statistical power of the experiment. In practice, budget constraints and other factors often limit the number of samples and replicates we can test. A complementary approach is to compare your data set with similar datasets from other sources to maximize the statistical power of the grouped data set. This is known as <b>meta-analysis</b>. Meta-analysis is particularly useful today because the number of omics datasets is very high compared to the past and for commonly studied systems, it's possible to have dozens if not hundreds of related datasets. However, meta-analysis is not trivial. In our single proteomics dataset, we identified one batch effect, but in meta-analysis, each dataset is likely to have its own unique batch effects. These can result from experimental design, sample prep strategy, instrument used to generate the data, the skills of the lab staff, geographical location, and numerous other sources. Because of the complexity of meta-analysis, many labs choose not to do this. When properly performed however, meta-analysis can provide an additional level of confidence that the biomarkers you have identified are biologically and statistically significant.
One common criticism of omics data is whether the results are real or a result of experimental artifacts. As a data set gets bigger, it becomes more likely that you will begin to find statistically significant results purely by chance. To forestall such criticisms, it is standard procedure to experimentally validate key omics results whenever possible. In our case, that means testing the potential biomarkers experimentally to prove that the patterns we see are actually happening. There are several ways to experimentally validate omics results, the most common of which are listed below:
| Omics Method | Experimental Validation Method |
| --- | --- |
| Transcriptomics | Quantitative PCR, Northern Blot |
| Epigenomics | ChIP, Protein-Binding Assay |
| Proteomics | Western Blot |
| Metabolomics/Lipidomics/Glycomics | HPLC, NMR |
Once validated with experimental results, we can begin comparing our potential biomarker to known biomarkers using the regression methods we have already discussed. More sophisticated methods of biomarker validation can be found in the scientific literature, and you should now have the background to study these methods on your own.
To summarize, this would be the strategy we would follow for identifying potential proteomic biomarkers:
1. Generate hypothesis designed to address your biological question
2. Design proteomics experiment with proper controls and identify relevant covariates
3. Generate proteomics data
4. Differential analysis of proteomics data to identify potential biomarkers indicating relevant state changes
5. Pathway analysis to link potential biomarkers to pathways relevant to your hypothesis and to identify differentially expressed pathways
6. Experimental validation of biologically relevant biomarkers using Western blots
7. Comparison of potential biomarkers to known biomarkers to assess clinical utility
## But Wait, There's More!
For a traditional bioinformatics analysis, this would be the end of this particular experiment. However, at the time of this writing, the field of bioinformatics is undergoing a rapid transition. We now have publicly available omics datasets from thousands of laboratories spanning 20+ years. We are also rapidly trasitioning to <b>single cell</b> and <b>spatial omics</b> methods which allow for much much higher resolution omics studies than traditionally available. This obviously represents a huge amount of data and analysis by traditional methods is becoming difficult. This is precipitating a transition of biomedical research to cloud environments coupled to <b>machine learning</b> techniques designed to handle massive, complex data sets. While this may sound daunting, we've already covered basic techniques involved in machine learning, including linear and logistics regression, principle components analysis, and clustering. While there are many applications for machine learning in biomedical data science, biomarker discovery is especially amenible to these methods and thus we will introduce basic ML models for biomarker discovery in the final submodule, <b>Submodule 9: Biomarker Discovery Using Machine Learning</b>.
---
<p><span style="font-size: 30px"><b>Quizzes</b></span> <span style="float : inline;">(run the command below to display the quizzes)</span> </p>
<code>
IRdisplay::display_html('<iframe src="quizes/Chapter8_Quizes.html" width=100% height=450></iframe>')
</code>
---
<code>
sessionInfo()
</code>
---
## Conclusion
This submodule demonstrated the identification of potential protein biomarkers for Ischemia Reperfusion Injury (IRI) using linear regression-based differential analysis with the `limma` package in R. By incorporating both treatment and time covariates, we revealed significant protein abundance changes between sham and placebo groups at the 6-hour mark, as well as alterations induced by treprostinil treatment. While this analysis provides a strong starting point, further steps are crucial for solidifying these findings. These include pathway analysis to assess biological relevance, potential meta-analysis with comparable datasets to enhance statistical power, and experimental validation using methods like Western blots. The next submodule will explore the application of machine learning techniques for biomarker discovery, offering a powerful complement to traditional bioinformatics approaches in the era of big data and increasingly complex experimental designs.
## Clean up
Remember to move to the next notebook or shut down your instance if you are finished.
<div style="display: flex; justify-content: center; margin-top: 20px; width: 100%;">
<div style="display: flex; justify-content: space-between; width: 50%;">
<div>
<a href=https://github.com/NIGMS/Analysis-of-Biomedical-Data-for-Biomarker-Discovery/blob/master/GoogleCloud/Submodule07_Exploratory_Proteomic_Analysis.ipynb#overview>Previous section</a>
</div>
<div>
<a href="#top8">Top of this page</a>
</div>
<div>
<a href=https://github.com/NIGMS/Analysis-of-Biomedical-Data-for-Biomarker-Discovery/blob/master/GoogleCloud/Submodule09_Biomarker_Discovery_using_ML.ipynb#overview>Next section</a>
</div>
</div>
</div>
## References
[Hou J, Tolbert E, Birkenbach M, Ghonem NS. Treprostinil alleviates hepatic mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed Pharmacother. 2021 Nov;143:112172. doi: 10.1016/j.biopha.2021.112172. Epub 2021 Sep 21. PMID: 34560548; PMCID: PMC8550798.][hou]<br>
[Ding M, Tolbert E, Birkenbach M, Gohh R, Akhlaghi F, Ghonem NS. Treprostinil reduces mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed Pharmacother. 2021 Sep;141:111912. doi: 10.1016/j.biopha.2021.111912. Epub 2021 Jul 15. PMID: 34328097; PMCID: PMC8429269.][ding]<br>
[ding]: https://pubmed.ncbi.nlm.nih.gov/34328097/ "Ding M, Tolbert E, Birkenbach M, Gohh R, Akhlaghi F, Ghonem NS. Treprostinil reduces mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed Pharmacother. 2021 Sep;141:111912. doi: 10.1016/j.biopha.2021.111912. Epub 2021 Jul 15. PMID: 34328097; PMCID: PMC8429269."
[hou]: https://pubmed.ncbi.nlm.nih.gov/34560548/ "Hou J, Tolbert E, Birkenbach M, Ghonem NS. Treprostinil alleviates hepatic mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed Pharmacother. 2021 Nov;143:112172. doi: 10.1016/j.biopha.2021.112172. Epub 2021 Sep 21. PMID: 34560548; PMCID: PMC8550798."
|
{
"filename": "Submodule08_Differential_Analysis_Proteomics_2.ipynb",
"repository": "NIGMS/Analysis-of-Biomedical-Data-for-Biomarker-Discovery",
"query": "transformed_from_existing",
"size": 50848,
"sha": ""
}
|
Subsets and Splits
No community queries yet
The top public SQL queries from the community will appear here once available.