
INSTRUCTION
stringlengths 48
14.1k
| RESPONSE
stringlengths 14
8.79k
| SOURCE
stringclasses 1
value | METADATA
stringlengths 48
55
|
|---|---|---|---|
In Vcf file, generate by samtools mpileup, there is an AD field (Allelic depth). This field is suppose to replace the previous DP4 field (Number of high-quality ref-forward, ref-reverse, alt-forward and alt-reverse bases).
But when I use -t DP4, AD in mpileup, I have a different number of values in the 2 fields:
GT:PL:DP:DP4:AD 0/1:27,0,107:5:4,0,1,0:4,1,0
And I don't understand why one data is missing in the AD field... Is it not supposed to be like the DP4, so:
> high-quality ref-forward, ref-reverse, alt-forward and alt-reverse bases
?
After that I am not sure: what there are 3 values in the AD field ? And what are the differences between AD and DP4?
Thanks for your help.
Rob | I think DP4 shows ref and all alt reads, but AD further separates the different alt alleles (a single record can have multiple alt alleles).
If you check the AD and DP4 descriptions in the VCF header, it should explain both fields.
I actually don't have AD in my samtools 1.3 VCF. They may have taken it out or it might depend on some other setting. Thus, I can't really confirm. | biostars | {"uid": 214307, "view_count": 4019, "vote_count": 2} |
Does anyone have a simple solution to downloading all the refseq genomes for a particular taxon?
Using `ncbi-genome-download` its possible to specify the species or genus TaxIDs and download them, but apparently you can't go higher up the taxonomic ranks (even though enterobacteria has a TaxID of 543 for instance).
If anyone knows of a way to download all the Enterobacteria I'm all ears.
Alternatively, if there is a method of extracting the species TaxIDs from the Enterobacterial taxid in NCBI such that I can pass them all directly to `ncbi-genome-download` that would work too. | From speaking with a few other pros, this was the solution in the end (though only very rough at the mo):
Use the `ete3` toolkit to get a list of IDs:
from ete3 import NCBITaxa
import sys
taxon_name = sys.argv[1]
ncbi = NCBITaxa()
ncbi.update_taxonomy_database()
ebact = ncbi.get_descendant_taxa(taxon_name)
with open('./taxids', 'w') as ofh:
for i in ebact:
ofh.write("%s\n" % i)
# At this point, one could import ncbi-genome-download as a python method and continue
Which gave me a list of IDs (though this includes ALL descendent taxa, even ones without complete genomes etc).
I passed these to the latest version of `ncbi-genome-download` which accepts a `--taxid 12345,65890` format for specifiying the IDs.
So I just ran:
for file in * ;
do python ~/bin/ncbi-genome-download/ncbi-genome-download-runner.py -l complete -v -p 10 --taxid $(paste -s -d ',' "$file") bacteria ;
done
I had to run this iteratively on many files after I split my `taxids` file up as there is a limit to how many args can be passed to `--taxid` at once.
EDIT Sept 2018:
I contributed a script to the `ncbi-genome-download` repo to make getting the TaxIDs nice and easy. It uses the approach above, but there’s no need to rewrite it for oneself now. | biostars | {"uid": 302533, "view_count": 2271, "vote_count": 1} |
Hi all,
I'm looking for a simple solution for renaming fasta headers.
I have this fasta header
>trpE___AA_HMM___6fa05435949258489b608db9e58e5ba38821f2f26fffe5755daff43abin_id:MALBOS1|source:AA_HMM|e_value:5.2e99|contig:MALBOS1_000000117228|gene_callers_id:113772|start:215745|stop:217260|length:1515
And I would like to rename it only like this
>MALBOS1_000000117228
That means, remove everything before the pattern "contig:" and after "|gene_callers_id"
Any ideas?
thanks
| perl -pe 's/>.+contig:(.+?)\|.+/>$1/' < FASTA_IN > FASTA_OUT | biostars | {"uid": 9546056, "view_count": 562, "vote_count": 1} |
Hi, all.
Can anybody tell help me explain which parent comes first when writing the genotype?
if phased,
mom 0/1 dad 0/0
child 1|0 or 0|1?
many thanks. | I am not working with trios myself - so I am just thinking out loud.
Since the child may have novel variants and the parents may also share variants, could we always correctly assign a variant to either parent? Many times would possible, but other times not.
I believe that the order `0|1` vs `1|0` has meaning only in terms of phasing. Every variant in a block lists the genotypes to be in sync with other variants in that block. That is the sole purpose of the order. For unphased variants, the order is always low to high `0/1`
To figure out which parent shares the variant look at the parent genotype. | biostars | {"uid": 9512398, "view_count": 391, "vote_count": 1} |
<p>I have gene lists that I want to compare based on how well they were conserved across human and mice. Is there an established way of doing this? My first impulse was to compare identity directly but since genes have differing lengths and differing functional sizes of differing importance, a measure just based on sequence identity seems like a bad way of comparing any two genes to each other.</p>
| <p>Hi oganm, you can do this the complicated way or the easy way. The easy way is with identities (from Ensembl BioMart for instance) and the complicated way could be with a BLASTP similarity network.</p>
<p>Yes orthologs have differing length but in a pairwise comparison (like human-mouse), you have limited info.</p>
| biostars | {"uid": 141131, "view_count": 4067, "vote_count": 1} |
I am trying to do exactly the same pipeline for RNA-seq data process as the TCGA does. Usually when we ask a sequencing service, we can get a fastQ file. It contains sequence and read quality information. The alignment step comes next.
However in the case of TCGA, as their pipeline suggested ([https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/][1]). It seems that they used BAM files as one of the inputs. I was wondering why they used BAM files as inputs and how can I repeat what they did?
In addition, why isn't there seem to be a adaptor trimming process?
[1]: ![TCGA mRNA-seq pipeline schematic][1]https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
[1]: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/images/gene-expression-quantification-pipeline-v3.png | https://gdc.cancer.gov/about-gdc/gdc-faqs - Read the answer to "How can I access GDC sequencing data in FASTQ format?"
Level 1 (aka the raw fastq) data is restricted; to request access to it, see instructions here: https://gdc.cancer.gov/access-data/obtaining-access-controlled-data
Adaptor trimming is unnecessary in RNA-seq read mapping; many papers have been written about it (e.g. https://academic.oup.com/nargab/article/2/3/lqaa068/5901066 ). | biostars | {"uid": 9506203, "view_count": 1045, "vote_count": 1} |
Dear All,
I have to deal some small molecules screening for my protein-drug docking studies. Please suggest me few small molecule databases ..also the free software can use to screen them...thank you | What are you actually trying to achieve? Your question is extremely vague.
Do you just want a big collection of drug-like molecule? Grab <a href="http://www.gdb.unibe.ch/gdb/home.html">GDB-17</a>
Do you want a curated source of biologically interesting molecules? Go to <a href="http://www.ebi.ac.uk/chebi/">ChEBI</a>
Do you want small molecules with annotated activity? Try <a href="https://www.ebi.ac.uk/chembldb/">ChEMBL</a>
Do you want small molecules bound to a protein? Try <a href="http://www.rcsb.org/pdb/home/home.do">PDB</a>
For docking, just go to any literature search engine and search for small molecule docking. You will get reviews, algorithms and papers describing software. There is plenty of literature, so start reading.
Edit: After several comments, it seems the interest lies in identifying any compound which may inhibit the activity of the given receptor. For this purpose I would suggest using <a href="http://zinc.docking.org/">ZINC</a>, which also has vendor information. | biostars | {"uid": 115992, "view_count": 5855, "vote_count": 1} |
I am trying to convert BAM files to Bigwig format. I was originally using a combination of genomeCoverageBed and BedgraphToBigWig to go from BAM to Bigwig in 2 steps:
samtools sort file.bam -o file.sorted.bam;
genomeCoverageBed -bg -split -ibam file.sorted.bam -g file.chrom.sizes > file.bedgraph;
sort -k1,1 -k2,2n file.bedgraph >file.sorted.bedgraph;
bedGraphToBigWig file.sorted.bedgraph file.chrom.sizes file.bw;
After finding out that deeptools (function called bamCoverage) could convert BAM to Bigwig AND normalize all in one go, I decided to use it. I first decided to not normalize (just for testing purposes)
bamCoverage -b file.bam -o file.bw
This command works but I can't help but notice the output file.bw in both scenarios are drastically different. My starting BAM file is 1.1GB. When using bedGraphToBigWig, my output file is 122MB. When using bamCoverage, it's 23MB.
I know that you can change the --binSize in bamCoverage which will lead to a larger file. I tried a bin of 10 and my file was 45MB.
My question is: what is the relationship between file sizes between the 2 software? I am confused as to why the size is so different. From what I understand, in bamCoverage, the coverage is calculated as the number of reads per bin, where bins are short consecutive counting windows of a defined size. But what is the equivalent setting in BedGraphtoBigwig? Is it -blockSize or maybe itemsPerSlot? Their defaults are 256 and 1024 respectively so I am a bit confused.
I am not sure whether this is something to worry about? Presumably, all it means is that my 2 files (the one produced with BedgraphToBigwig and the one produced with bamCoverage) will have different 'resolutions', when visualized on a something like IGV or UCSC Genome Browser. Is this correct?
| The reason is that `genomeCoverageBed` by default spills out bedGraphs at the base-pair level, so it piles up the depth for every base. In contrast, `bamCoverage` has a default 50bp window (option `-bs`, see documentation) that it aggregates reads over. I always use `-bs 1` because visually these tracks looks much nicer/smoother than the default `-bs 50`. | biostars | {"uid": 333364, "view_count": 3979, "vote_count": 2} |
<p>Hi. I just have a quick question in regards to calculating the N50 for a list of contigs. Anybody know how to do it in excel, by any chance? I'm not much of a programmer just yet, and I was just wondering if there was an easier way than just constructing a python program or something.</p>
<p>Thanks!</p>
| Get the lengths of your contigs into a column, and sort that column. Sum over all lengths to get the total number bases N, and then just traverse your list of lengths from top to bottom until the cumulative sum reaches N/2. (You could use an additional column to keep track of the running sum if that makes it easier.)
The length of the contig where your running sum passes N/2 is the N50. (In the case where the running sum exactly equals N/2, then the N50 is the average of this length and the subsequent one in the list.)
For example, below, the cumulative sum exceeds 44/2=22 on a contig with length 6, so N50 = 6.
contigs length cumulative sum
AAAAAAA 7 7
AAAAAA 6 13
AAAAAA 6 19
AAAAAA 6 25
AAAA 4 29
AAAA 4 33
AAA 3 36
AAA 3 39
AA 2 41
AA 2 43
A 1 44 | biostars | {"uid": 134275, "view_count": 10444, "vote_count": 1} |
Dear all,
referring to the batch correction methods for scRNA-seq, would you have any preference and/or comments ? among possible choices :
-- MNNCorrect, as outlined in SimpleSingleCell workflows :
https://bioconductor.org/packages/release/workflows/html/simpleSingleCell.html
-- ZINB-WAVE :
https://bioconductor.org/packages/release/bioc/html/zinbwave.html
-- HARMONY :
https://www.biorxiv.org/content/10.1101/461954v2
-- SCTransform :
https://satijalab.org/seurat/v3.0/integration.html
thanks a lot,
bogdan | The results seem to be very experiment-specific. For example, in today's [SCRIBE pre-print][1], all the methods (except the one introduced) perform poorly:
![enter image description here][2]
One thing to notice is that they all fail in different ways, so the problems don't seem to be due to some artifact in the data itself. For example, MNN mixes NF and TH, but Seurat splits PEP.
[1]: https://www.biorxiv.org/content/10.1101/793463v2
[2]: https://user-images.githubusercontent.com/6363505/66263486-70484680-e7c1-11e9-99ee-a0cb9bcd05db.png | biostars | {"uid": 401404, "view_count": 4644, "vote_count": 3} |
I have a pipeline script called `pipeline.sh` . I usually execute this for a single sample like so:
$ pipeline.sh sample1 sample1.R1.fastq.gz sample1.R2.fastq.gz
Where $1 is sample ID, $2 and $3 are the read files.
I use GNU parallel with a parameters file that specifies the paths to each file.
$ nohup parallel -j 4 -a params.pipeline.txt --colsep '\s+\ ./pipeline.sh
I want to automate my pipeline so that I don't need a parameters file and it looks through the folders for the reads (same file structure as they come out of the sequencer).
I have:
parallel -j 4 ./pipeline.sh {/1.} ::: *.R1.fastq.gz :::+ *.R2.fastqgz
However this assumes the fastqs are in the same folder, how can I change my parallel so that it searches through the folder structure for the files.
| using nextflow (not tested, but it should look like this):
nextflow.enable.dsl = 1
params.directories=""
process scanDirectories {
output:
path("paths.txt") into paths
script:
"""
find ${params.directories} -type f -name "*.R1.fq.gz" \
awk -F '/' '{S=\$NF;gsub("\\.R1\\.fq\\.gz\$","",S);F2=\$0;gsub("\\.R1\\.fq\\.gz\$",".R2.fq.gz",F2);printf("%s,%s,%s\\n",S,\$0,F2);}' > paths.txt
"""
}
paths.splitCsv(header: false,sep:',',strip:true).set{pipe_in}
process runPipeline {
tag "${sample}"
input:
tuple val(sample),val(R1),val(R2) from pipe_in
output:
path("result.txt") into result_ch
script:
"""
echo "DO Something ${sample} ${R1} ${R2}" > result.txt
"""
}
and then something like
nextflow run -resume script.nf --directories "/path/to/dir1 /path/to/dir2"
| biostars | {"uid": 9526066, "view_count": 541, "vote_count": 1} |
I designed a bait set to caputure genes but i would like to know whether my baits bind on other places other than the designed gene sequences. My idea is to blast it back to the reference genome and identify the "background noise" of the non gene sequences. An easy solution i think is to remove all gene sequences from my reference genome. Leaving only the intergenic and repetitive regions.
Does someone have a easy solution for this?
Or other suggestion i could try? | get a BED for your genes and then use bedtools mask fasta http://bedtools.readthedocs.io/en/latest/content/tools/maskfasta.html to replace the bases with N. | biostars | {"uid": 312585, "view_count": 1048, "vote_count": 1} |
<p>Does Pindel generate genotypes or does it only report on sites? Adding extra characters to get to 80.</p>
| There is a `pindel2vcf` binary inside Pindel package to convert Pindel calls to genotyped vcf. | biostars | {"uid": 124174, "view_count": 1672, "vote_count": 1} |
I am trying to use the Bingo Plugin in Cytoscape but the output are empty and I can not figure out why. Can you guys give me some enlightment? Here is a screenshot of my input.
More one question: is 'Reference set' the background of genes?
Thank you guys.
![< image not found >][1]
[1]: http://imageshack.com/a/img661/9255/aHtdnw.png | I've not used it for a while, but be sure your Identifiers(Ensemble I guess in this case) can be mapped to the ontology files. It is better to update the ontology and annotation files in Bingo before using it. | biostars | {"uid": 123044, "view_count": 2263, "vote_count": 1} |
Hi all,
I'm looking to analyze mouse ERVs from bulk-RNAseq but have mostly found data resources and pipelines for human ([ERVmap][1], [HERVd][2]). I've also found a mouse [ERE database][3] that appears to no longer be supported.
The only reasonable resource I've found for mouse ERVs is [gEVE][4] but wanted to ask if anyone is aware of additional resources?
[1]: https://www.pnas.org/content/115/50/12565
[2]: https://herv.img.cas.cz/
[3]: https://www.ncbi.nlm.nih.gov/pubmed/22691267
[4]: https://academic.oup.com/database/article/doi/10.1093/database/baw087/2630466 | Can you be more specific regarding what resources you're after?
You might have better luck with gEVE:
https://academic.oup.com/database/article/doi/10.1093/database/baw087/2630466 | biostars | {"uid": 405475, "view_count": 1012, "vote_count": 1} |
This post is related to, but different (a continuation) from my earlier post at https://www.biostars.org/p/268060/ seeking help with running the genome assembler software - ABySS. I got my ABySS runs going.
But in order to assess the optimal k-mer, I was trying values 21-97 (increments of 2).
Related to this attempt, at k-mer value>64 (i.e. 65), the STDERR log terminated with the line - "ABYSS: ../Common/Kmer.h:48: static void Kmer::setLength(unsigned int): Assertion `length <= 64' failed."
Obviously my university's compute cluster has ABySS compiled and configured to allow k-mers <= 64.
Some context for my questions below - I have 2*150 PE Illumina HiSeq400 reads from fungal spore DNA (haploid genome). Read length trimming has resulted in size distribution between 41 (min) and 150 (max).
1a. How do I execute ABySS with k-mer value > 64, but WITHOUT re-compiling? Is that possible? I ask in the context of what is written at https://insidedna.me/tool_page_assets/pdf_manual/abyss.pdf, which reads:
> ./configure --enable-maxk=96
>
> "The default maximum k-mer size is 64 and may be decreased to reduce
> memory usage or increased at compile time. This value must be a
> multiple of 32 (i.e. 32, 64, 96, 128, etc):"
1b. If running k-mer > 64 is NOT possible without recompiling, is there a work-around that allows re-setting local variable(s) on just my compute cluster account?
2. Is there a theoretical / practical limit on k-mer value > default 64? I know SPAdes, for example, permits as high as 127. What is it for ABySS? Is it 128?
3. Is checking k-mer value > 64 **necessary**? Which is a different question from whether it is **possible**?
Thank you!
| Hi Anand,
Glad to hear you were able to get some assemblies running!
To answer your questions:
* Unfortunately, there is no way to run an assembly with k-mer size > 64 without re-compiling the software.
* There is no upper bound for `--enable-maxk`. However: (i) increasing `--enable-maxk` increases the memory requirements of ABySS, and (ii) you will have to adjust some MPI settings for large k-mer sizes, as described here: https://github.com/bcgsc/abyss/wiki/ABySS-Users-FAQ#2-my-abyss-assembly-jobs-hang-when-i-run-them-with-high-k-values-eg-k250
* Yes, it is very likely that you will need to use k-mer sizes > 64 in order to achieve the best possible assembly of your data
If you are uncomfortable with compiling software from source, you can perhaps get your IT team to do it for you (if you have an IT team). That said, knowing how to compile software from source is a very useful skill in the bioinformatics domain. If you are interested in learning, here is an introductory tutorial that may be helpful: http://www.thegeekstuff.com/2012/06/install-from-source/ | biostars | {"uid": 269480, "view_count": 2948, "vote_count": 1} |
<p>I've been working on editing some code on the server at my work, I've come into a problem where we need a snp quality to go through our filters downstream in the pipeline that we use. Does anyone know if Snp Quality exists in the VCF file? If so where? </p>
| <p>SNP Quality can be represented in several places in a VCF file. </p>
<p>1) the QUAL column, which is the phred-scaled quality score for the assertion made in ALT. In other words, it's: 10log_10 prob(call in ALT is wrong). </p>
<p>2) GQ, encoded in the FORMAT column is genotype quality, encoded as a phred score: -10log_10p(genotype call is wrong). </p>
<p>3) Especially if you're looking at tumor/normal pairs, you may see that it's represented as VAQ (variant quality).</p>
<p>The header of the VCF should give a description of exactly which fields are present in your files and help you determine which ones contain the quality scores that you're looking for.</p>
<p>For more info, check out the <a href='http://www.1000genomes.org/node/101'>VCF format description</a></p>
| biostars | {"uid": 9897, "view_count": 31062, "vote_count": 8} |
I just want to make sure my interpretation is correct: a MAPQ value of 255 indicates that the mapping quality is "not available" because it's a unique alignment, and a unique alignment means that it has a 0 probability of mapping to the wrong place, therefore: -10*log10(0 probability of error) = inf (ie. "not available")
| tldr: MAPQ of 255 has little actual meaning.
Whether 255 actually means that an alignment is unique or "I don't want to bother returning a meaningful value" is aligner dependent. In practice, tools that produce MAPQ of 255 are doing so because they aren't calculating a real MAPQ and are just indicating, "this is a more likely alignment than anything else I could find". How much more likely this particular alignment is is purely a matter of the settings you use, which are pretty arbitrary. As a rule of thumb, assume that MAPQ values from splice-aware aligners lack any standard phred-scale interpretation. | biostars | {"uid": 372666, "view_count": 4796, "vote_count": 1} |
<p>I'm very new to bioinformatics so any suggestion how to start ?</p>
<p>my field is microbiology & molecular biology</p>
| Hi!,
I have been there! Do you know any computer programming or scripting language! If not, start with a Linux or awk tutorial. There are several very user friendly bioinformatics tools. Install them into your computer, choose a project and try to analyze some sample data. Get some NGS data try to analyze it. If you do not have it yourself, try to download from publications or sample data from Illumina website. Start with simple things such as check sequence base quality, genome coverage, estimation of insert size, etc Following three bioinformatics tools will give you a good start: bedtools, samtools, bamtools. You can check official documentation for installation, if you are using a mac computer, here is a page explaining how to install them: http://genescripts.com/
Just try to use each command and figure out what they doing. You will learn a lot. Visit forums like this and read them. Try to take free courses online from Coursera, Mitedx, Udacity, etc. A great public tool is [Galaxy project](http://galaxyproject.org/). There are many tutorial video there. You will learn a lot. Have fun during your studies! | biostars | {"uid": 98983, "view_count": 7726, "vote_count": 2} |
Hi, currently, I am trying to calculate the coverage for my genes of interest (i.e. number of reads falling in or overlapping by the gene of interest) from DNA-seq data in a BAM file. Genes of interest are annotated in a BED file.
There are several [answers][1] on Biostar suggesting to use
`bedtools coverage` e.g., `bedtools coverage -abam sample.bam -b exons.bed -counts`. However, while checking the Samtools' manual, I stumbled upon `samtools bedcov` with the following description:
> read depth per BED region. Reports read depth per genomic region, as specified in the supplied BED file.
which can be used like this: `samtools bedcov gene.bed sample.bam`.
I have tested these two and I am getting different results! For instance, for `OR4F5`gene I get the following results (using the same BAM file for both):
chr1 69091 70008 "OR4F5" 61 #from bedtools coverage
and
chr1 69091 70008 "OR4F5" 4714 #from samtools bedcov
where `61` is very different than `4714`!
Another observation is that the result from `Samtools` is instantaneous for the above test while `Bedtools` takes a lot of time to produce the result. The speed itself has led me to think of using `samtools bedcov`. However, I was wondering whether there is catch that I am overlooking! I tried to find more information about `samtools bedcov` but I was not able to find anything more than that one-liner description.
I would be happy to hear about your feedback on this.
**[EDIT]:** This is how it looks like in IGV. It seems that `Bedtools` results is closer to reality.
![visualization in the IGV][2]
[1]: https://www.biostars.org/p/61748/
[2]: http://oi66.tinypic.com/2uztpue.jpg | The description that goes with `samtools bedcov` is wrong (I'll create an issue on github). It returns the sum of per-base coverage in each region. I'm not sure how that would be useful, but that's what it is.
**Edit:** [Here's the issue](https://github.com/samtools/samtools/issues/588) to get the description fixed. | biostars | {"uid": 195497, "view_count": 23689, "vote_count": 11} |
Disclaimer: Tried to post this on bioconductor support but it wont allow me. I tried adding an entire paragraph in "English language" but no - still wouldn't allow me.
Hi everyone,
I am using DEXSeq for exon quantification. I ran dexseq_prepare_annotation to convert gencode v24 GTF to GFF like this:
python2.7 ~/path/to/R/library/DEXSeq/python_scripts/dexseq_prepare_annotation.py gencode.v23.annotation.gtf gencode.v23.annotation.gff
For IDO2 which has gene id ENSG00000188676, I got 18 exonic parts in the GFF:
grep 'ENSG00000188676' gencode.v23.annotation.gff
chr8 dexseq_prepare_annotation.py aggregate_gene 39934614 40016391 . + . gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39934614 39934954 . + . transcripts "ENST00000343295.8"; exonic_part_number "001"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39934955 39935218 . + . transcripts "ENST00000343295.8+ENST00000502986.2"; exonic_part_number "002"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39949149 39949165 . + . transcripts "ENST00000343295.8+ENST00000502986.2"; exonic_part_number "003"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39949166 39949264 . + . transcripts "ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "004"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39963608 39963703 . + . transcripts "ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "005"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39979067 39979186 . + . transcripts "ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "006"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39982652 39982770 . + . transcripts "ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "007"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39984951 39985507 . + . transcripts "ENST00000343295.8"; exonic_part_number "008"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39985508 39985522 . + . transcripts "ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "009"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39985523 39986460 . + . transcripts "ENST00000343295.8"; exonic_part_number "010"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39986900 39987085 . + . transcripts "ENST00000343295.8"; exonic_part_number "011"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39987743 39987870 . + . transcripts "ENST00000418094.1"; exonic_part_number "012"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39987871 39987970 . + . transcripts "ENST00000418094.1+ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "013"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 39989721 39989838 . + . transcripts "ENST00000418094.1+ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "014"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 40005327 40005378 . + . transcripts "ENST00000389060.8+ENST00000502986.2"; exonic_part_number "015"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 40013565 40013713 . + . transcripts "ENST00000418094.1+ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "016"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 40015247 40015605 . + . transcripts "ENST00000418094.1+ENST00000343295.8+ENST00000389060.8+ENST00000502986.2"; exonic_part_number "017"; gene_id "ENSG00000188676.13"
chr8 dexseq_prepare_annotation.py exonic_part 40015606 40016391 . + . transcripts "ENST00000418094.1+ENST00000343295.8+ENST00000502986.2"; exonic_part_number "018"; gene_id "ENSG00000188676.13"
However, when I go to [UCSC genome browser][1], it shows that IDO2 has 10 exons. Why does my GFF show 18 exonic parts or is there an issue with the conversion?
[1]: https://genome.ucsc.edu/cgi-bin/hgGene?hgg_gene=uc064mgi.1&hgg_prot=ENST00000389060.8&hgg_chrom=chr8&hgg_start=39949165&hgg_end=40015605&hgg_type=knownGene&db=hg38&hgsid=609990501_JhxAO5bBRafZjnld9AXbEMaABB6e | An "exonic part" is simply a part of an exon, so there will be at least as many of them as there are exons. Take the following example of a gene with two isoforms:
####--------####----
####------####----##
Here, `#` is an exon and `-` is intronic or intergenic region. I'll merge all of those exons together and then illustrate where the exonic parts are:
####------######--##
1111------223344--55 (1-5 indicate that the base above belongs to that `exonic part`)
You can see that the exons are divided into disjoint sections, where each "part" is shared completely by all of the transcripts that contain it (compare exon 2 in both of the isoforms, which are only partially shared between them).
| biostars | {"uid": 274809, "view_count": 2176, "vote_count": 2} |
Hello guys,
It may seem like a basic question, but this is causing confusion.
What is the difference between fold change and Log fold change?
Regards,
| If you think that fold-change is the 'expression level' in one set of samples (set A) divided by the 'expression level' in another set (set B), then log-fold-change is the log of that value (typically to base 2).
That is, if FC=, A/B, then log_FC = log(A / B) = log(A) - log(B)
and if log_FC=x, then FC=2^x | biostars | {"uid": 312980, "view_count": 33880, "vote_count": 7} |
Hello,
I am analysing RNA-seq data to investigate differential gene expression in hybrids compared to parental species. Since I work with natural populations, I have few samples (5 of two different tissues for each species and hybrid).
I am using the DESEQ2 package for my expression analysis. What I observe is that many genes, although they have a high log fold change (more than 1 or even 1.5), they have a padj > 0.1. While this is true in one group, in another group, genes with a log fold change of 1 or even lower are having a padj < 0.1 or even padj < 0.05.
I was wondering what are the reasons for this observation?
Thank you in advance. | Fold changes tend to be higher when genes have overall lower expression (which means low counts).
Since low counts have lower power than high counts the significance for these fold changes is often low unless these FCs are supported by many replicates.
Example 1: Two genes had expression of 50 an 5. That would be a fold change of 10.
Example 2: Two genes had expression of 5000 an 500. That would be a fold change of 10 as well.
Still, the second one is much more reliable as the first one could be a product of the technical noise produced by the sequencing. Adding or reducing e.g. 10 counts to example a can change the result quite much:
`50 - 10` vs `5 + 5` would already change the original FC from 10 to 2.6 whereas
`5000 - 10` - `500 + 5` changes the FC from 10 to 9.88.
You can see that higher counts are less affected by small fluctuations in counts, therefore they are more reliable.
In DESeq2 you can check the `baseMean` column to get the average expression. This is probably low for many of these genes with high FCs but large padj. You can visualize this relationship of baseMean to logFC with the `plotMA` function.
This is where the concept of shrinkage kicks in. It aims to estimate the "true" fold changes from the data. As you can see below there is little evidence for the fold changes of the genes with low baseMean to be actually true, so they are shrunken towards zero. If you want lowly-expressed genes to be significant 8given they in fact are DEG) then you need most importantly many replicates and high sequencing depth.
Check the DESeq2 vignette for it.
Some examples:
Unshrunken FCs:
![enter image description here][1]
Shrunken:
![enter image description here][2]
[1]: https://i.imgur.com/8nsORxP.png
[2]: https://i.imgur.com/ufNK0sH.png | biostars | {"uid": 435466, "view_count": 1362, "vote_count": 1} |
Does anyone know of a simple scripting solution to take a list of accessions and pull out the gene name from the header of FASTA sequences?
For instance given the accession: XP_016469325.1
and given the FASTA entry:
>XP_016469325.1 Nicotiana tabacum|C3H|C3H family protein
MEEELLKRNTDCVYFLASPLTCKKGIECEYRHSEIARLNPRDCWYWLAESCLNPTCAFRH
PPLESHAETSSESAPPQHKSAVPVNKTNVPCYFYFNGYCIKGERCSFLHGPDDGTTTWKS
SKIASGVPDGPTAEKKTSVGSETGPASVEKPSNSSETGSKAAAHEYIKSQVDLISMTNDV
GEQSASHETSGSPSEEATAVRLDSLVPAEGFTQGGSDLSPDWSSDEEVEDNVEREEWLES
SPGFDVLVDDRIEGWSHKDDHSYLLQHDRECDERFAGYDFENNLEYDPAYPDMRIVSDEE
LDDSYYSKVESHEVNEYAREIVIPAHGRQSIPHKRKFPREPGFCARGNVDLRDLLKKRRV
IESDPPNYLSRRLDLSRFNAREQCRDRHRPQGSRWMPQSLASKLESNSSFSSGFVDATRL
EGANQLKKLRQSHRSSYRQQHFKDRRRGRSQPFANETPRRMASRQRSTEVPKIFGGPKTL
AQIREEKIKGREDGNSFERTVPSGGSEREDFSGPKPLSEILKDKRRLSSVVNFSN
I would like to have output the gene name "C3H" that is spanned by the "|"
This script I modified from a previous post can grab the gene names, however I'm not sure how to only get the gene names corresponding to a separate list of accessions (accessions.list).
with open('PlantTFDB_ALL_TF_pep.fas','r') as f:
for line in f:
if '>' in line:
line = line.strip().split('|')
print(line[1]) | File `acc.txt` contains the accession IDs:
$ cat acc.txt
XP_016469325.1
Firstly, getting the mapping relationship between acc and gene name and saving them to acc2gene.tsv
$ grep '>' seqs.fa | \
perl -ne 'next unless />(\S+).+\|(.+?)\|/; print "$1\t$2\n";'
XP_016469325.1 C3H
Secondly, joining acc.txt and acc2gene.tsv using csvtk (http://bioinf.shenwei.me/csvtk/download/) or other tools:
$ csvtk join -H -t -k acc.txt acc2gene.tsv | csvtk cut -H -t -f 2
<hr/>
Original answer
Searching header line using `grep -f `, and capturing accession ID and gene name using regular expression in Perl.
$ grep '>' seqs.fa | \
grep -f acc.txt | \
perl -ne 'next unless />(\S+).+\|(.+?)\|/; print "$1\t$2\n";'
XP_016469325.1 C3H
If you just want the gene names, that would be easier:
$ grep '>' seqs.fa | grep -f acc.txt | cut -d '|' -f 2
C3H
| biostars | {"uid": 272376, "view_count": 6243, "vote_count": 1} |
Hi there!
I´m using KIRC database from TCGA and downloaded FPKM values, then converted to TPM with this formulas:
FPKMtoTPM <- function(x) {
return(exp(log(x) - log(sum(x)) + log(1e6)))
}
df <- data %>%
mutate_if(is.numeric, FPKMtoTPM)
Now i want to convert TPM to log2(TPM+1). How can i do this?
Thanks!
David | Just as you defined the first function FPKMtoTPM you can define another one for the log transformation of the TPM values
logTPM <- function(x) {return(log2(x+1))}
df %>% mutate_if(is.numeric, logTPM)
| biostars | {"uid": 9479234, "view_count": 3672, "vote_count": 2} |
Hi All,
I tried to download some BS-Seq data from [McGill Epigenomics Mapping Centre (\[Here Database Link\]\[1\])][1]
I have a little assumption about the BS-Seq wig/bigwig files in http://epigenomesportal.ca/edcc/ database.
KNIH wig file is 0-10 scale, DEEP/CEEHRC wig files are 0-100 scale while blueprint and Roadmap are 0-1 scale. Anyone met this problem before?
It looks these wig file are coverage not methylation signal. the scale actually represent the sequence depth.
Thanks.
[1]: http://edcc-dev2.udes.genap.ca/cgi-bin/directory.cgi?b=2017-10&as=1&i=3&ctc=1&session | **I obtained a response from IHEC and it seems my assumption is right, each dataset is uploaded by different group.**
The annotation tracks deposited on the IHEC Data Portal have been generated by each respective data producing consortia, each with their own softwares and parameters. This explains the difference you see across tracks of different groups. IHEC is currently working on generating a pan-IHEC dataset using a unified data analysis pipeline and standardized quality control metrics. It will however take several more months before we reach this step. If you want to do real comparison across IHEC members datasets, I would currently recommend obtaining the raw data. | biostars | {"uid": 331073, "view_count": 970, "vote_count": 2} |
Hello. I've been trying to use awk to solve the following problem for some time with no luck. Was hoping someone here could give me some clues as to what I'm doing wrong.
I have a file that looks like this:
INPUT:
>chr8:76290516-76290880
578 T
579 G
580 A
>chr14:22131464-22132025
468 T
469 G
470 A
>chr12:33695439-33695441
468 T
469 G
470 A
Each record in the file has a header that starts with `>` I would like to print a new column which is essentially a line number, starting after the header which starts with `>`, and to begin counting from the number following the `:` in the header. I would like each record (after the header `>`) in the file to be treated/counted independently.
I have tried to do this in steps, starting with adding the counts to the third column first, and then will attempt to add the header value to the counts following that. I have had no luck with getting the counts in the third column using this command below:
awk '{FS = "/n"}{RS = ">"}{if(!/^>/){print $1, $2, NF, $3 }}' input.txt
DESIRED OUTPUT:
>chr8:76290516-76290518
578 T 76290516
579 G 76290517
580 A 76290518
>chr14:22131464-22131466
468 T 22131464
469 G 22131465
470 A 22131466
>chr12:33695439-33695441
321 T 33695439
322 G 33695440
333 A 33695441
Any pointers would be very much appreciated. Thank you so much!!
EDIT: I did not include my failed attempt to solve this problem, as @mensur pointed out to me, so I have edited to include. Thank you. | awk -F '[:]' '/^>/ {print;P=$2;X=0;next;} {print $0, (P+X);X++;}' input.txt | biostars | {"uid": 9474902, "view_count": 1160, "vote_count": 1} |
Hi everyone !!
I tried to install Zinba, a tool for peak calling used in the analysis of ATAC-seq datas.
- Operating system that I am using : Linux (computing cluster) and R (version 3.4.3)
- Error message : compilation aborted for bed2vector.cc (code 2)
make: *** [bed2vector.o] Error 2
ERROR: compilation failed for package ‘zinba’
- Command used : install.packages("zinba_2.02.03.tar.gz", repos=NULL)
But I've also tried to install zinba_2.03.1.tar.gz and zinba_2.02.04.tar.gz and I have the same error message.
- Copy of the output :
>install.packages("zinba_2.03.1.tar.gz")
Installing package into ‘/sulb2/amontois/R/x86_64-pc-linux-gnu-library/3.4’
(as ‘lib’ is unspecified)
inferring 'repos = NULL' from 'pkgs'
* installing *source* package ‘zinba’ ...
** libs
icc -I/apps/brussel/CO7/magnycours-ib/software/R/3.4.3-intel-2017b-X11-20171023/lib64/R/include -DNDEBUG -I/apps/brussel/CO7/magnycours-ib/software/imkl/2017.3.196-iimpi-2017b/mkl/include -I/apps/brussel/CO7/magnycours-ib/software/X11/20171023-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/Mesa/17.2.4-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libGLU/9.0.0-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/cairo/1.14.10-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libreadline/7.0-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/ncurses/6.0-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/bzip2/1.0.6-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/XZ/5.2.3-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/zlib/1.2.11-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/SQLite/3.20.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/PCRE/8.41-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libpng/1.6.32-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libjpeg-turbo/1.5.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/LibTIFF/4.0.8-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/Java/1.8.0_152/include -I/apps/brussel/CO7/magnycours-ib/software/Tcl/8.6.7-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/Tk/8.6.7-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/cURL/7.56.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libxml2/2.9.4-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/GDAL/2.2.2-intel-2017b-Python-2.7.14/include -I/apps/brussel/CO7/magnycours-ib/software/PROJ/4.9.3-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/GMP/6.1.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/NLopt/2.4.2-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/FFTW/3.3.6-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libsndfile/1.0.28-GCCcore-6.4.0/include -fpic -O2 -xHost -ftz -fp-speculation=safe -fp-model source -c aliType.c -o aliType.o
ire/SQLite/3.20.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/PCRE/8.41-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libpng/1.6.32-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libjpeg-turbo/1.5.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/LibTIFF/4.0.8-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/Java/1.8.0_152/include -I/apps/brussel/CO7/magnycours-ib/software/Tcl/8.6.7-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/Tk/8.6.7-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/cURL/7.56.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libxml2/2.9.4-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/GDAL/2.2.2-intel-2017b-Python-2.7.14/include -I/apps/brussel/CO7/magnycours-ib/software/PROJ/4.9.3-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/GMP/6.1.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/NLopt/2.4.2-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/FFTW/3.3.6-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libsndfile/1.0.28-GCCcore-6.4.0/include -fpic -O2 -xHost -ftz -fp-speculation=safe -fp-model source -c alignAdjust.cc -o alignAdjust.o
[...]
icpc -I/apps/brussel/CO7/magnycours-ib/software/R/3.4.3-intel-2017b-X11-20171023/lib64/R/include -DNDEBUG -I/apps/brussel/CO7/magnycours-ib/software/imkl/2017.3.196-iimpi-2017b/mkl/include -I/apps/brussel/CO7/magnycours-ib/software/X11/20171023-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/Mesa/17.2.4-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libGLU/9.0.0-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/cairo/1.14.10-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libreadline/7.0-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/ncurses/6.0-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/bzip2/1.0.6-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/XZ/5.2.3-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/zlib/1.2.11-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/SQLite/3.20.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/PCRE/8.41-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libpng/1.6.32-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libjpeg-turbo/1.5.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/LibTIFF/4.0.8-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/Java/1.8.0_152/include -I/apps/brussel/CO7/magnycours-ib/software/Tcl/8.6.7-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/Tk/8.6.7-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/cURL/7.56.1-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/libxml2/2.9.4-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/GDAL/2.2.2-intel-2017b-Python-2.7.14/include -I/apps/brussel/CO7/magnycours-ib/software/PROJ/4.9.3-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/GMP/6.1.2-GCCcore-6.4.0/include -I/apps/brussel/CO7/magnycours-ib/software/NLopt/2.4.2-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/FFTW/3.3.6-intel-2017b/include -I/apps/brussel/CO7/magnycours-ib/software/libsndfile/1.0.28-GCCcore-6.4.0/include -fpic -O2 -xHost -ftz -fp-speculation=safe -fp-model source -c bed2vector.cc -o bed2vector.o
In file included from /gpfs/software/CO7/magnycours-ib/software/GCCcore/6.4.0/bin/../include/c++/6.4.0/ext/hash_set(60),
from pc.h(5),
from bed2vector.cc(1):
/gpfs/software/CO7/magnycours-ib/software/GCCcore/6.4.0/bin/../include/c++/6.4.0/backward/backward_warning.h(32): warning #1224: #warning directive: This file includes at least one deprecated or antiquated header which may be removed without further notice at a future date. Please use a non-deprecated interface with equivalent functionality instead. For a listing of replacement headers and interfaces, consult the file backward_warning.h. To disable this warning use -Wno-deprecated.
#warning \
^
bed2vector.cc(41): error: "hash" is ambiguous
hash_map<string, int, hash<string>,equal_to<string> > cind_map;
^
bed2vector.cc(111): error: "hash" is ambiguous
hash_map<string, int, hash<string>,equal_to<string> >::const_iterator li=cind_map.find(chr);
^
compilation aborted for bed2vector.cc (code 2)
make: *** [bed2vector.o] Error 2
ERROR: compilation failed for package ‘zinba’
* removing ‘/sulb2/amontois/R/x86_64-pc-linux-gnu-library/3.4/zinba’
Warning message:
In install.packages("zinba_2.03.1.tar.gz") :
installation of package ‘zinba_2.03.1.tar.gz’ had non-zero exit status
- It seems to have an error with the installation of bed2vector... But I don't really understand what's wrong...
Can you help me to solve this problem of installation please ??
Thank you in advance !
Anais | The ZINBA code was written before C++11, which is when `hash` was added to the standard library, and so the authors used the g++ extension instead. Based on your output, the compiler is using C++11 (or later), so it has two versions of the class `hash` available and can't figure out which to use.
Quick Fix
---
You can temporarily direct the compiler to use a specific C++ version for installation using `withr::makevars`. Try
library(withr)
with_makevars(c(CXXFLAGS = "-std=gnu++98"), install.packages("zinba_2.03.1.tar.gz", repos = NULL))
Long Game
---
I hit different errors than you on both OS X (clang) and Linux (g++), so expect to hack on the code to get it working ([I started to last year](https://github.com/mfansler/zinba), but had other priorities). A more permanent workaround for your issue is to untar/zip the archive and add a **Makevars** file to the `zinba/src` directory containing the line:
PKG_CXXFLAGS = -std=gnu++98
Leave the source unzipped and install with `install.packages("your/path/to/zinba", repos = NULL, type = "source")`. | biostars | {"uid": 299405, "view_count": 2017, "vote_count": 1} |
I have a text file with 4 columns: gi, evalue, start, stop.
There's several duplicates of each gi but they're not identical in coverage, so I want to extract from the text file only certain rows: those HSPs or hits (this a tblastn output) which have the greatest coverage for that gi.
HOWEVER, gi isn't enough of a determinant, because there is often more than one 'top' hsp per genome/gi. That is, there is more than one copy of the gene in a genome. Evalue doesn't seem to be enough a decider here in any scenario so column 2 can be ignored. Once the hits for a particular gi are distinguished based on their genome positions, I will simply take the hit which has the longest coverage (stop-start).
What I would like to do is in this intermediate step is, per gi entry (e.g. below), identify separate groups of hits/hsps based on their start/stop positions, or rather employ some sort of hierarchal clustering to set the rows in groups which could for instance be denoted in a 5th "ID" column, values 1,2,3 etc.
I don't yet know how to go about it (most of the clustering literature out there seems to go beyond what I need) but for instance, if the first row in a particular gi group was assigned ID 1, if the other rows in that group had start and stop values that differed by more than 600 nt say to those of this one, they would belong to a separate group (i.e. impose a cut-off 600). And so on, with hits not belonging to one used to nucleate other subgroup (2,3 etc.).
I started to employ numpy and generated a difference array for all start values with all other start values, and the same for stop, but the scripting to go through each row and call on this array was going to get very convoluted. I know scipy has some clustering algorithms but I can't tell if that's applicable to my problem.
If there is a simpler route you can think of please let me know.
Below is an extract of the data, where column gi is an extract for all values for that particular gi entry. In this entry there is an additional column showing sequence lengths.
```
99036121 0.0 1392057 1390123 1934
99036121 0.0 1392099 1390123 1976
99036121 0.0 1392111 1390123 1988
99036121 0.0 1392123 1390123 2000
99036121 0.0 1392123 1390543 1580
99036121 0.0 1730139 1728823 1316
99036121 0.0 1730139 1728829 1310
99036121 0.0 1768983 1767775 1208
99036121 1e-133 1768950 1767778 1172
99036121 1e-69 1768983 1768216 767
99036121 1e-77 1390509 1390123 386
99036121 1e-83 1768983 1767787 1196
99036121 2e-117 1768563 1767775 788
99036121 2e-58 1768983 1768279 704
99036121 3e-121 1768950 1767775 1175
99036121 3e-123 1768950 1767775 1175
99036121 3e-133 1768983 1767775 1208
99036121 3e-93 1768428 1767775 653
99036121 4e-101 1768950 1767775 1175
99036121 4e-135 1768983 1767775 1208
99036121 4e-136 1768983 1767775 1208
99036121 5e-133 1768983 1767787 1196
99036121 5e-136 1768983 1767775 1208
99036121 6e-112 1768542 1767775 767
99036121 6e-138 1768983 1767775 1208
99036121 7e-96 1768533 1767775 758
99036121 8e-136 1768983 1767775 1208
``` | This is straightforward with Bioconductor GRanges. The process is roughly:
- Load data for each GI
- Create GRanges for each HSP
- Use `reduce()` to collapse all overlapping hits into contiguous blocks
The return value, gr1, contains the three contiguous blocks of hits.
```r
z = read.table(textConnection("99036121 0.0 1392057 1390123 1934
99036121 0.0 1392099 1390123 1976
99036121 0.0 1392111 1390123 1988
99036121 0.0 1392123 1390123 2000
99036121 0.0 1392123 1390543 1580
99036121 0.0 1730139 1728823 1316
99036121 0.0 1730139 1728829 1310
99036121 0.0 1768983 1767775 1208
99036121 1e-133 1768950 1767778 1172
99036121 1e-69 1768983 1768216 767
99036121 1e-77 1390509 1390123 386
99036121 1e-83 1768983 1767787 1196
99036121 2e-117 1768563 1767775 788
99036121 2e-58 1768983 1768279 704
99036121 3e-121 1768950 1767775 1175
99036121 3e-123 1768950 1767775 1175
99036121 3e-133 1768983 1767775 1208
99036121 3e-93 1768428 1767775 653
99036121 4e-101 1768950 1767775 1175
99036121 4e-135 1768983 1767775 1208
99036121 4e-136 1768983 1767775 1208
99036121 5e-133 1768983 1767787 1196
99036121 5e-136 1768983 1767775 1208
99036121 6e-112 1768542 1767775 767
99036121 6e-138 1768983 1767775 1208
99036121 7e-96 1768533 1767775 758
99036121 8e-136 1768983 1767775 1208
"),header=FALSE)
> library(GenomicRanges)
> # Note that start and end are switched in these example data
> # so you may need to be careful in the next line if this is
> # not generally true.
> gr = GRanges(seqnames=z[,1],ranges=IRanges(start=z[,4],end=z[,3]))
> gr1 = reduce(gr)
> gr1
GRanges object with 3 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] 99036121 [1390123, 1392123] *
[2] 99036121 [1728823, 1730139] *
[3] 99036121 [1767775, 1768983] *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
``` | biostars | {"uid": 142937, "view_count": 2098, "vote_count": 1} |
Dear all,
I've got a large list of variant as VCF format per chromosome (human), which annotated using VEP (release 98) as offline mode. But, I didn't get the corresponding rsid at the output. Could you please help me out how I can obtain the "rsid" during the annotation?
Thanks | [--check_existing=1](http://www.ensembl.org/info/docs/tools/vep/script/vep_options.html#existing)
> Checks for the existence of known variants that are co-located with
> your input. By default the alleles are compared and variants on an
> allele-specific basis - to compare only coordinates, use
> --no_check_alleles.
>
> Some databases may contain variants with unknown (null) alleles and
> these are included by default; to exclude them use
> --exclude_null_alleles.
>
> See this page for more details.
>
> Not used by default | biostars | {"uid": 415490, "view_count": 2464, "vote_count": 3} |
Hi everyone, I'm performing analysis of some RNAseq samples, and currently trying to cope with batch effect.plotting PCA of vsd transformed data, I can clearly see two batches which are differ fromt the others.
plotPCA(vsd, intgroup = c('batch'),ntop= 34085)
![PCA][1]
If I'm not mistaken, then for DE expression analysis I could use design formula of dds function, to reduce batch effect:
dds <- DESeqDataSetFromMatrix(countData = counts, colData = coldata, design =~ batch + type)
dds <- DESeq(dds)
vsd <- varianceStabilizingTransformation(dds, blind = TRUE)
But I also would like to visualise results of batch correction. For PCA plot I could simply follow the DESeq2 manual and just perform limma batch effect removal:
mat <- assay(vsd)
mat <- limma::removeBatchEffect(mat, vsd$batch)
assay(vsd) <- mat
plotPCA(vsd, intgroup = c('batch'),ntop= 34085)
Which gives me a nice results:
![batch corrected PCA][2]
But now I'd like to visualise expression of individual genes of interest. I could use vsd transforemd data, but these numbers mask the actual count numbers, therefore laking info on actual level of gene expression, which I'd like to preserve.
![expression of gene X fo given conditions, vsd transformed data, limma correction][3]
So what would be the better solution here? Could I use limma removeBatchEffect() on normalised count table, maybe?
Thanks!
[1]: https://i.ibb.co/bzspVz1/Intitial-pca.png
[2]: https://i.ibb.co/RQhzx2x/batch-corrected-PCA.png
[3]: https://i.ibb.co/Cs6jD0j/geneX.png | `vst` transforms to approximately log2-scale, that is simply a data transformation to allow a wide range of counts to fit on a narrow scale rather than having counts spread between 0 and numbers in the hundreds-of- thousands. | biostars | {"uid": 380030, "view_count": 3084, "vote_count": 1} |
I am attempting to learn a bit more about processing RNA-seq data, and I am starting off by trying to recreate some figures from a paper. To do this I need to import the data from GEO. They have stored they data in SubSeries and SuperSeries and I have been attempting to download the SubSeries and use the data. I initially tried importing that data into R using:
mRNA_data <- getGEO('GSE42379', GSEMatrix=TRUE)
When looking at the data, it downloaded into two separate GSE series. I have been unable to access the data from both of those GSE files, as it only seems to parse the first of the files. Is this because the data was obtained on two different platforms? How would I be able to access the data in the second GSE series? The only way I have found that will access all the data is if you use `GSEMatrix=FALSE` which parses all 28 samples but then to access them is a complete nightmare, which again is not all that well explained (or at least to my limited abilities/understanding). The data I am trying to access is [here][1].
Thanks
[1]: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE42379 | GEOquery always returns a `list` when using `GSEMatrix = TRUE` for exactly situations like this where the Series record has data from multiple platforms.
```
library(GEOquery)
mRNA_data <- getGEO('GSE42379', GSEMatrix=TRUE)
names(miRNA_data)
length(miRNA_data)
# mRNA_data is a `list`
human_data = mRNA_data[["GPL8759"]]
mouse_data = mRNA_data[["GPL8321"]]
human_data
```
Now, `human_data` and `mouse_data` are each `ExpressionSet` objects. | biostars | {"uid": 305749, "view_count": 2518, "vote_count": 1} |
I have a nextflow workflow for which I am running with sge + apptainer profile using the command `nextflow run main.nf -profile sge,apptainer`
but I am receiving the error `line #: bwa: command not found`
I have tried running `apptainer run workflow.sif` to check, and `bwa` as well as other tools seem to be properly installed. I am not sure why nextflow is not finding the tools...
The `workflow.sif` file is built by running `apptainer build workflow.sif Apptainer`
The Apptainer file to build the .sif is:
Bootstrap: docker
From: rocker/r-ubuntu:22.04
%post
# automake
apt-get update \
&& apt-get install -y --no-install-recommends build-essential automake bzip2 wget unzip \
python3 python3-dev python3-pip python3-venv git git-lfs default-jdk ant \
libbz2-dev libsdl1.2-dev liblzma-dev libcurl4-openssl-dev zlib1g-dev libxml2-dev \
r-cran-tidyverse bwa samtools multiqc datamash && rm -rf /var/lib/apt/lists/*
# CONDA
%environment
export LC_ALL=C
export LC_NUMERIC=en_GB.UTF-8
export PATH="/opt/miniconda/bin:$PATH"
%post
#essential stuff but minimal
apt update
#for security fixe:
#apt upgrade -y
apt install -y wget bzip2
#install conda
cd /opt
rm -fr miniconda
#miniconda3: get miniconda3 version 4.7.12
wget https://repo.continuum.io/miniconda/Miniconda3-4.7.12-Linux-x86_64.sh -O miniconda.sh
#install conda
bash miniconda.sh -b -p /opt/miniconda
export PATH="/opt/miniconda/bin:$PATH"
#add channels
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
#install trimmomatic
conda install -y -c conda-forge -c bioconda nextflow
conda install -y -c conda-forge -c bioconda trimmomatic
conda install -y -c conda-forge -c bioconda gatk4
conda install -y -c conda-forge -c bioconda fastqc
#cleanup
conda clean -y --all
rm -f /opt/miniconda.sh
apt autoremove --purge
apt clean
# RSTUDIO
mkdir -p /usr/local/lib/R/etc/ /usr/lib/R/etc/
echo "options(repos = c(CRAN = 'https://cran.rstudio.com/'), download.file.method = 'libcurl', Ncpus = 4)" | tee /usr/local/lib/R/etc/Rprofile.site | tee /usr/lib/R/etc/Rprofile.site
R -e 'install.packages("remotes")'
# Update apt-get
Rscript -e 'install.packages("remotes", version = "2.4.2")'
Rscript -e 'remotes::install_cran("rmarkdown",upgrade="never", version = "2.19")'
Rscript -e 'remotes::install_cran("knitr",upgrade="never", version = "1.41")'
Rscript -e 'remotes::install_cran("tidyverse",upgrade="never", version = "1.3.2")'
Rscript -e 'remotes::install_cran("plotly",upgrade="never", version = "4.10.1")'
Rscript -e 'remotes::install_cran("RColorBrewer",upgrade="never", version = "1.1-3")'
Rscript -e 'remotes::install_cran("data.table",upgrade="never", version = "1.14.6")'
Rscript -e 'remotes::install_cran("viridis",upgrade="never", version = "0.6.2")'
Rscript -e 'remotes::install_cran("DT",upgrade="never", version = "0.26")'
%runscript
exec /bin/bash "$@"
%startscript
exec /bin/bash "$@"
The nextflow.config is:
params {
...
max_memory = 10.GB
max_cpus = 4
max_time = '48.h'
}
process {
withLabel: big_mem {
cpus = "${params.max_cpus}"
memory = "${params.max_memory}"
time = "${params.max_time}"
penv = 'smp'
}
}
profiles {
conda {
conda.enabled = true
docker.enabled = false
apptainer.enabled = false
process.conda = "./envs/env.yml"
}
mamba {
conda.enabled = true
conda.useMamba = true
docker.enabled = false
apptainer.enabled = false
}
docker {
conda.enabled = false
docker.enabled = true
docker.userEmulation = true
apptainer.enabled = false
process.container = "directory/myworkflow:latest"
}
apptainer {
conda.enabled = false
apptainer.enabled = true
apptainer.autoMounts = true
docker.enabled = false
process.container = 'file://myworkflow.sif'
}
sge {
process {
executor = "sge"
scratch = true
stageInMode = "copy"
stageOutMode = "move"
errorStrategy = "retry"
clusterOptions = '-S /bin/bash -o job.log -e job.err'
}
executor {
queueSize = 1000
}
}
}
manifest {
name = 'directory/myworkflow'
homePage = 'https://github.com/directory/myworkflow'
description = 'analysis pipeline'
mainScript = 'main.nf'
nextflowVersion = '!>=22.10.0'
version = '1.1.0'
}
env {
PYTHONNOUSERSITE = 1
R_PROFILE_USER = "/.Rprofile"
R_ENVIRON_USER = "/.Renviron"
}
// keep trace
trace {
enabled = true
file = "${params.outdir}/trace.txt"
overwrite = true
}
// keep report
report {
enabled = true
file = "${params.outdir}/report.html"
overwrite = true
}
// Function to ensure that resource requirements don't go beyond
// a maximum limit
def check_max(obj, type) {
if (type == 'memory') {
try {
if (obj.compareTo(params.max_memory as nextflow.util.MemoryUnit) == 1)
return params.max_memory as nextflow.util.MemoryUnit
else
return obj
} catch (all) {
println " ### ERROR ### Max memory '${params.max_memory}' is not valid! Using default value: $obj"
return obj
}
} else if (type == 'time') {
try {
if (obj.compareTo(params.max_time as nextflow.util.Duration) == 1)
return params.max_time as nextflow.util.Duration
else
return obj
} catch (all) {
println " ### ERROR ### Max time '${params.max_time}' is not valid! Using default value: $obj"
return obj
}
} else if (type == 'cpus') {
try {
return Math.min( obj, params.max_cpus as int )
} catch (all) {
println " ### ERROR ### Max cpus '${params.max_cpus}' is not valid! Using default value: $obj"
return obj
}
}
}
| What is your Nextflow version? Edit: Ah now I see it, > 22.10.0, but based on https://github.com/nextflow-io/nextflow/issues/2970 dedicated Apptainer support has just been added recently. Try `NXF_VER=22.11.0-edge nextflow run ...`. Alternatively, switch back to just using Singularity, that works for sure, I use it routinely. | biostars | {"uid": 9556018, "view_count": 740, "vote_count": 2} |
Hi Senior Methylators:
This figure is derived from fastqc analysis to a BS-seq fastq data.
However, Why the content of "G" is lowest rather than "C"?
OK. Maybe I get the idea: it is from Sample_2.fastq?
<img alt="image" src="https://lh3.googleusercontent.com/-Shw0GF_tvfM/Vkg1q9LgKlI/AAAAAAAAAIA/mhBpviAITjY/w346-h250/407F.tm.png" style="height:250px; width:346px" />
| <p>Yup, you nailed it, it's because you're looking at sample #2, where you'll have a lack of Gs and an abundance of As.</p>
| biostars | {"uid": 165825, "view_count": 2248, "vote_count": 3} |
I have 10x sc RNA-seq data for which I want to do an RNA velocity analysis using scvelo and CellRank. However, the bottleneck for me is generating the spliced/unspliced counts from the aligned data (BAM files or 10x output). I've found two tools that are able to do this:
(1) [Velocyto][1]. The original tool for calculating spliced/unspliced counts. The CLI tool is simple to use and has an option `velocyto run10x` which should work directly on 10x data. One potential issue with velocyto is that is seems to be no longer updated. The last commits to the git repository are from ~3 years ago (as of 07/2021).
(2) [STARsolo][2]. The updated version of STAR has a built-in tool to calculate spliced/unspliced ratios. Basically adding `--soloFeatures Gene Velocyto` as option should produce this result. The issue is that the output has a different form (spliced.mtx and unspliced.mtx files) from the .loom files required for scvelo.
Hence neither of the tools is working for me, even after quite some troubleshooting (a few of the error files attached).
**(1) How does one convert the output of STARsolo into a form that can be read in python by scvelo (e.g. a loom file like velocyto does)?**
**(2) Are there are other tools out there, that can generate splicing data for sc-RNAseq data from 10x, which are directly compatible with scvelo?**
=====================**Error logs**============================================
**Velocyto run 10x**
I created a conda environment with velocyto and all its dependencies and simply ran
``velocyto run10x -vvv --dtype uint32 $DATA10x_PATH $GTF_PATH``.
``OMP: Info #270: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
2021-07-06 18:17:21,016 - ERROR - This is an older version of cellranger, cannot check if the output are ready, make sure of this yourself
2021-07-06 18:17:21,016 - ERROR - Can not locate the barcodes.tsv file!
Traceback (most recent call last):
File "/Users/*/opt/miniconda3/envs2/velocyto/bin/velocyto", line 11, in <module>
sys.exit(cli())
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/click/core.py", line 1137, in __call__
return self.main(*args, **kwargs)
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/click/core.py", line 1062, in main
rv = self.invoke(ctx)
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/click/core.py", line 1668, in invoke
return _process_result(sub_ctx.command.invoke(sub_ctx))
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/click/core.py", line 1404, in invoke
return ctx.invoke(self.callback, **ctx.params)
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/click/core.py", line 763, in invoke
return __callback(*args, **kwargs)
File "/Users/*/opt/miniconda3/envs2/velocyto/lib/python3.9/site-packages/velocyto/commands/run10x.py", line 91, in run10x
bcfile = bcmatches[0]
IndexError: list index out of range``
The 10x output (barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz) are in subfolder ``count/sample_feature_bc_matrix``, rather than ``outs/filtered_gene_bc_matrices`` as required by velocyto. However, renaming these folders does not solve the problem.
==============**Velocyto run**=========
I also tried running velocyto directly on the BAM files, similar to ``velocyto run -b filtered_barcodes.tsv -o output_path -m repeat_msk_srt.gtf possorted_genome_bam.bam mm10_annotation.gtf``. However, here I get a Permission error with ``samtools``.
==============**STARsolo** ===========
This gets more complicated with more options, but I was able to run
``STAR --genomeDir $GENOME_PATH \
--runThreadN 12 \
--readFilesIn $FASTQ_FILES_1 $FASTQ_FILES_2 \
--outFileNamePrefix $RESULTS_PATH \
--outSAMtype BAM SortedByCoordinate \
--outSAMattributes Standard \
--outFilterType BySJout \
--outFilterMultimapNmax 1 \
--outFilterMismatchNmax 1 \
--outFilterIntronMotifs RemoveNoncanonical \
--outSJfilterOverhangMin 30 20 20 20 \
--outSJfilterCountUniqueMin 3 2 2 2 \
--clipAdapterType CellRanger4 --outFilterScoreMin 30 \
--soloType CB_UMI_Simple \
--soloFeatures Gene Velocyto \
--soloMultiMappers Unique \
--soloCBwhitelist $CB_WHITE_LIST \
--soloUMIlen 12 \
--soloCBmatchWLtype 1MM --soloUMIfiltering MultiGeneUMI_All --soloUMIdedup 1MM_CR \
--soloCellFilter EmptyDrops_CR 3000 0.99 10 45000 90000 5000 0.01 20000 0.01 10000``
without problems and generate most of the alignment required. It generated a separate folder for the velocyto files, with files ``ambiguous.mtx`` ``barcodes.tsv`` ``features.tsv`` ``spliced.mtx`` ``unspliced.mtx``. However, as mentioned I had problems working with these files in scvelo.
[1]: https://velocyto.org/velocyto.py/index.html
[2]: https://github.com/alexdobin/STAR/blob/master/docs/STARsolo.md | If you have the fastq files available you can use Alevin as described in their tutorial: https://combine-lab.github.io/alevin-tutorial/2020/alevin-velocity/ | biostars | {"uid": 9479044, "view_count": 4119, "vote_count": 1} |
I'm trying to carry out an eQTL analysis on some TCGA BRCA data I have.
For the genotypes, I have a list of birdseed files, that each have a hybridisation reference that looks like: `WRIED_p_TCGA_143_147_150_Hahn_N_GenomeWideSNP_6_A09_799960`.
My RNA-Seq data is labelled with a portion of the sample barcodes, e.g., **TCGA.3C.AAAU** (which would correspond to the barcode **TCGA-3C-AAAU-01A-11R-A41B-07**).
For eQTL analysis, I need to pair up a genotype with the corresponding expression values, but I need a file providing the mappings. As per [the TCGA Website][1], I feel like its probably a SDRF file I need, but I'm not entirely sure about this, and I can't find one through the TCGA data portal in any case.
Could someone lend me some guidance?
[1]: https://wiki.nci.nih.gov/display/TCGA/Sample+and+Data+Relationship+Format | See if either of these help.
sdrf from Broad: https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/brca/cgcc/broad.mit.edu/genome_wide_snp_6/snp/broad.mit.edu_BRCA.Genome_Wide_SNP_6.mage-tab.1.2023.0/broad.mit.edu_BRCA.Genome_Wide_SNP_6.sdrf.txt
sdrf from UNC: https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/brca/cgcc/unc.edu/illuminahiseq_totalrnaseqv2/totalrnaseqv2/unc.edu_BRCA.IlluminaHiSeq_TotalRNASeqV2.mage-tab.1.1.0/unc.edu_BRCA.IlluminaHiSeq_TotalRNASeqV2.1.1.0.sdrf.txt | biostars | {"uid": 151169, "view_count": 3327, "vote_count": 1} |
In Seurat's pbmc3k tutorial, they set the CreateSeuratObject with various parameters including `min.cell=3` and `min.features=200`
Can anybody explain exactly what these two parameters mean and where they pull the numbers 3 and 200?
| The [Seurat manual](https://cran.r-project.org/web/packages/Seurat/Seurat.pdf) does a good job explaining the parameters for any function.
> **min.cells** Include features detected in at least this many cells. Will subset the counts matrix as well. To reintroduce excluded
> features, create a new object with a lower cutoff.
>
> **min.features** Include cells where at least this many features are detected.
The values they picked here are somewhat arbitrary, but `min.cells` helps limit the number of genes used by removing those unlikely to play any part in differentiating groups of cells due to being expressed in very few cells. In general, most genes removed will be those with zero counts across all cells. `min.features` removes dead cells cells and empty droplets where few genes are detected. | biostars | {"uid": 407339, "view_count": 7170, "vote_count": 2} |
<p>I am trying to get tophat / cufflinks to work with Ensembl annotations. </p>
<p>Although this looks like a long post, my problem is quite simple: I keep getting <strong>CUFF.1</strong>, <strong>CUFF.2</strong>, <strong>CUFF.3</strong>, etc, as my gene<em>id, in the genes.fpkm</em>tracking file:</p>
<pre><code>$ head genes.fpkm_tracking
tracking_id class_code nearest_ref_id gene_id gene_short_name tss_id locus length coverage FPKM FPKM_conf_lo FPKM_conf_hi FPKM_status
CUFF.1 - - CUFF.1 - - chr1:568966-570302 - - 1080.24 1019.13 1141.36 OK
CUFF.4 - - CUFF.4 - - chr1:979748-982084 - - 19.74 10.1647 29.3153 OK
CUFF.2 - - CUFF.2 - - chr1:982297-984413 - - 11.6648 5.19434 18.1353 OK
CUFF.3 - - CUFF.3 - - chr1:881909-892399 - - 24.6448 15.3299 33.9596 OK
</code></pre>
<p>What I want is to have the Ensembl ENSG annotations, and possibly gene_name as well.</p>
<p>Yes, I realize the chromosome annotation (1st column) in the GTF/GFF file must match the FASTA header exactly. </p>
<p>I have tried the "native" Ensembl GTF file (chromosomes numbered 1, 2, 3..., X, Y, MT) and a bowtie2 genome index built from Ensembl's GRCh37.69 FASTA:</p>
<pre><code>1 processed_transcript exon 11869 12227 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "1"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002234944";
1 processed_transcript exon 12613 12721 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "2"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002867822";
1 processed_transcript exon 13221 14409 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "3"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002312635";
</code></pre>
<p>I have tried the native GTF with "chr" added in, and with the FASTA file modified in the same way (and of course bt2 index rebuilt):</p>
<pre><code>chr1 processed_transcript exon 11869 12227 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "1"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002234944";
chr1 processed_transcript exon 12613 12721 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "2"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002867822";
chr1 processed_transcript exon 13221 14409 . + . gene_id "ENSG00000223972"; transcript_id "ENST00000456328"; exon_number "3"; gene_name "DDX11L1"; gene_biotype "pseudogene"; transcript_name "DDX11L1-002"; exon_id "ENSE00002312635";
</code></pre>
<p>I have also tried using cufflinks gffread utility, to make a GFF file from the above GTF file:</p>
<pre><code>chr1 processed_transcript gene 11869 14412 . + . ID=ENSG00000223972;Name=DDX11L1;transcripts=ENST00000450305,ENST00000456328,ENST00000515242,ENST00000518655
chr1 processed_transcript misc_RNA 11869 14409 . + . ID=ENST00000456328;Parent=ENSG00000223972;geneID=ENSG00000223972;gene_name=DDX11L1;Name=DDX11L1-002;type=pseudogene
chr1 processed_transcript exon 11869 12227 . + . Parent=ENST00000456328
chr1 processed_transcript exon 12613 12721 . + . Parent=ENST00000456328
chr1 processed_transcript exon 13221 14409 . + . Parent=ENST00000456328
</code></pre>
<p>For which the corresponding bowtie2 index has these entries:</p>
<pre><code>$ bowtie2-inspect -n /hulk/genomes/Ensembl/Homo_sapiens.GRCh37.69.dna.chromosome
chr10
chr11
chr12
chr13
...
</code></pre>
<p>I have even tried using the iGenomes files, with the same results!</p>
<p>The way I am running tophat and cufflinks is like this:</p>
<pre><code>tophat -G Homo_sapiens.GRCh37.69.gtf --transcriptome-index=Homo_sapiens.GRCh37.69.genes -p 24 -o output Homo_sapiens.GRCh37.69.dna Sample1_1.fq.gz Sample1_2.fq.gz
cufflinks -p 6 -o output output/accepted_hits.bam
</code></pre>
<p>It is almost like I am running it without annotations, but the tophat / cufflinks programs never complain about being unable to match annotations with the FASTA file or anything:</p>
<pre><code>[2012-11-09 17:11:40] Beginning TopHat run (v2.0.6)
-----------------------------------------------
[2012-11-09 17:11:40] Checking for Bowtie
Bowtie version: 2.0.2.0
[2012-11-09 17:11:40] Checking for Samtools
Samtools version: 0.1.18.0
[2012-11-09 17:11:41] Checking for Bowtie index files
[2012-11-09 17:11:41] Checking for reference FASTA file
[2012-11-09 17:11:41] Generating SAM header for /hulk/genomes/Ensembl/Homo_sapiens.GRCh37.69.dna.chromosome
format: fastq
quality scale: phred33 (default)
[2012-11-09 17:11:44] Reading known junctions from GTF file
[2012-11-09 17:12:02] Preparing reads
left reads: min. length=30, max. length=101, 999911 kept reads (89 discarded)
right reads: min. length=30, max. length=101, 999341 kept reads (659 discarded)
[2012-11-09 17:12:43] Creating transcriptome data files..
...snip...
[2012-11-09 18:10:58] Reporting output tracks
-----------------------------------------------
[2012-11-09 18:16:25] Run complete: 01:04:44 elapsed
You are using Cufflinks v2.0.2, which is the most recent release.
[18:16:26] Inspecting reads and determining fragment length distribution.
> Processed 120637 loci. [*************************] 100%
> Map Properties:
> Normalized Map Mass: 985482.42
> Raw Map Mass: 985482.42
> Fragment Length Distribution: Empirical (learned)
> Estimated Mean: 163.12
> Estimated Std Dev: 50.10
[18:17:15] Assembling transcripts and estimating abundances.
> Processed 120951 loci. [*************************] 100%
</code></pre>
<p>Yes, this topic has been discussed online ad nauseum. <a href="http://seqanswers.com/forums/archive/index.php/t-12694.html">Here</a>, and <a href="http://cufflinks.cbcb.umd.edu/gff.html">here</a>, and <a href="http://www.biostars.org/p/52016/">here</a>, and many other places. I feel like I have read every possible internet page about this topic, but still the problem vexes me. In the past I once had this working just fine. In fact, I found that tophat / cufflinks worked very nicely with Ensembl annotations. However, it seems like something has changed -- either the software or the annotations? Am I the only one experiencing this? Can someone help me see what I am overlooking? Any help would be much appreciated.</p>
<p>Here are the software versions I am using:</p>
<pre><code>$ bowtie2 --version
/usr/local/bin/bowtie2-2.0.2/bowtie2-align version 2.0.2
64-bit
Built on igm1
Wed Oct 31 23:16:47 EDT 2012
$ tophat --version
TopHat v2.0.6
$ cufflinks
cufflinks v2.0.2
linked against Boost version 104900
</code></pre>
| <p>It doesn't appear that you're setting either the -G/--GTF or the -g/--GTF-guide flags when you run cufflinks. See comments above as well.</p>
| biostars | {"uid": 56784, "view_count": 13204, "vote_count": 3} |
Hi all,
I have BioNano Scaffolds of sequence data (no solid reference at the moment) and I am trying to orient them in the correct way to so I can do PCR and attempt to complete the sequence. One of my scaffolds is 67 MB, but I believe (based on BLATs and other context clues from the project) that this sequence is actually reversed. Is there a way using samtools or the command line to easily flip this sequence? Given that it is so big I would rather stay on the cluster and not have to download it to my computer to try and flip - but if that is what I need to do so be it.
Thanks so much! | If you have the emboss package installed on the server you can use `revseq seq.fa -reverse -nocomplement -outseq seq.rev.fa`. | biostars | {"uid": 9484965, "view_count": 1106, "vote_count": 1} |
hi everyone,
I have just started learning genomics as a part of my bioinformatics degree and I've been introduced to linux for handling fastq files and using fast qc. can anybody suggest some good learning resources which is more inclined towards linux for genomics. | This [**UNIX primer**][1] from Korf lab is an excellent resource to get started.
Titus Brown's [**NGS data analysis workshop**][2] is also a great resource.
[1]: http://korflab.ucdavis.edu/Unix_and_Perl/current.html#part1
[2]: https://angus.readthedocs.io/en/2019/toc.html | biostars | {"uid": 9485624, "view_count": 1269, "vote_count": 7} |
Hi all.
Recently, I got sequence for H3K27ac ChIP-seq data performed in various stimulation and knockdown condition. I have processed the reads and did peak calling using macs2. Looking into IGV browser, the enrichment looks good and the peaks are identified. The peaks called are of in the size range from ~1kb to 10kb. My concern here is whether the size of peaks identifies is really broad or is it ok to get large size peaks from H3k27ac chip. Next if want to perform motif identification, should I take all the broad peaks or perform peak calling to get broad peaks of lesser size (~1kb).
I would highly appreciate your suggestions and feedbacks
Thanks
peak calling method:
ls bowtie_out/*.25M.bam | cut -d'/' -f2 | parallel --verbose 'macs2 callpeak -t bowtie_out/{} -c ../Input_R1.bam -g mm -n {=s/.25M.bam//g=} --verbose 2 --broad --broad-cutoff 0.01 --cutoff-analysis --fe-cutoff 4 --outdir macs_peak'
IGV browser snapshot:
<a href="https://ibb.co/GVRQ23r"><img src="https://i.ibb.co/2jd3tYp/Screenshot-from-2019-09-14-10-26-01-png.png" alt="Screenshot-from-2019-09-14-10-26-01-png" border="0"></a>
Zoom of portion highlighted in above image:
<a href="https://ibb.co/Yhchj2t"><img src="https://i.ibb.co/KW2Wy69/image10.png" alt="image10" border="0"></a><br /><a target='_blank' href='https://imgbb.com/'>free image hosting</a><br />
| If the signals in IGV are looking reasonable over background for ~10kb domains, it is totally acceptable. There are something called **Super-enhancers** which are minimum of 10-12kb in size with high H3K27ac signal. Just google the term, you'll find number of papers solely based on super-enhancers.
Regarding motif analysis, instead of using total peak length, identify nucleosome free region within your H3K27ac peaks. These are potentially identified by small valleys which lack signal within peaks. `Homer` tool has a function called `nfr` which is what you want.
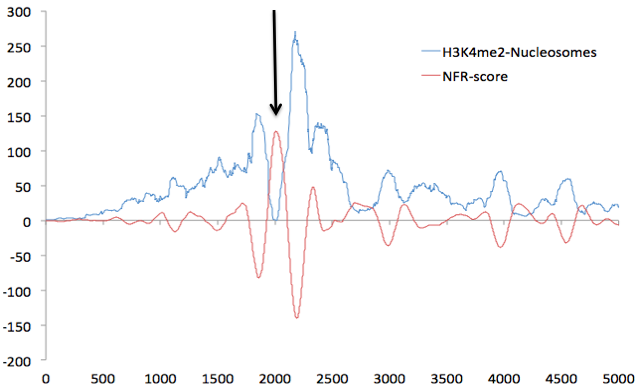
BTW, is there a specific reason to use `broad` option with MACS in your case? In my experience, narrowpeaks from MACS are of good quality and works very well for all types of analyses.
| biostars | {"uid": 398705, "view_count": 2527, "vote_count": 3} |
This is not a very memory friendly way of counting sequences from a multi fasta, any ideas to improve this?
generator = SeqIO.parse("test_fasta.fasta","fasta")
sizes = [len(rec) for rec in SeqIO.parse("test_fasta.fasta", "fasta")]
I'm avoiding using tools like grep since I want to make this more portable
| Standard Python will be faster than BioPython:
fh = open("test_fasta.fasta")
n = 0
for line in fh:
if line.startswith(">"):
n += 1
fh.close()
or shorter and possibly faster:
num = len([1 for line in open("test_fasta.fasta") if line.startswith(">")])
| biostars | {"uid": 294849, "view_count": 14413, "vote_count": 2} |
Hi Biostars
I am trying to convert sra from [PRJNA282735][1] dataset to fastq and I am getting following error...
> fastq-dump.2.1.7 fatal: SIGNAL - Segmentation fault
My fastq-dump command is
fastq-dump --split-3 SRR2016445.sra -O SRR2016445
I am not able to find similar error elsewhere. The [ENA page][2] for some samples of this dataset has three files per SRX experiment (e.g. SRR2016445.fastq, SRR2016445_1.fastq and SRR2016445_2.fastq).
This is unusual for me as I usually get one or two SRR runs per experiment (depending on single end paired end) but never 3. I am wondering if this is the reason for getting errors.
Anybody with similar experience?
[1]: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA282735/
[2]: http://www.ebi.ac.uk/ena/data/view/PRJNA282735 | I think 2.1. is fairly old, with a slightly newer version 2.4.2 I get:
fastq-dump.2.4.2 err: error unexpected while resolving tree within virtual file system module - failed to resolve accession 'SRR2016445' - Obsolete software. See https://github.com/ncbi/sra-tools/wiki ( 406 )
The latest release on github is 2.8, you should get and install the latest version as outlined here: https://github.com/ncbi/sra-tools/wiki | biostars | {"uid": 226446, "view_count": 8998, "vote_count": 1} |
Dear All,
I would like to ask you about an issue am facing. While doing exome sequencing I earlier did an estimation to what extent my reads were falling on the target regions, ie the target intervals that are used for target enrichment and I found for my samples the reads were around 75% spanning the exonic region. But now when am translating them into the variants and trying to find the somatic variants and annotating the somatic variants with all 3 annovar, oncotator and snpEff I find only 30% SNPs(novel ones which are not in dbSNP) are actually on the exons. The rest are spanning the intronic,intergenic,splice ,UTRs etc. Is this a likely scenario? How often do you see the SNPs mostly annotated in the non-exonic regions even when you use the exome sequencing provided your reads have high coverage in the exonic intervals used for target enrichment? I would like to know your advice in such cases.
Thanks | Does this include synonymous mutations? If you filtered out synonymous exonic mutations you'll find that is a large contributor as well. Most of the non-exonic regions you listed are all either targeted, or will be captured due to overlap (some areas extending into introns) and the majority of all mutations in a cell happen to occur in non exonic regions simply because exonic regions make up such a small percentage of the genome. Of course since you have filtered out off-target variants in this case, you will enrich for exonic variants.
Coupled with filtering out mutations that are also in dbSNP (which many valid somatic mutations may overlap or match identically mutations found in dbSNP by the way, plenty of known somatic variants that appear in COSMIC, TCGA, etc are also found in dbSNP) the number you are reporting (30%) doesn't surprise me that much. | biostars | {"uid": 110862, "view_count": 3279, "vote_count": 1} |
I am trying to calculate the coverage of a ChIP data set over a very limited set of gene coordinates. I have very simple bed files for both (after trying to eliminate any cause of the error) that are just the bare bones "chr start end" columns, and every time I run the command:
bedtools coverage -a a.bed -b b.bed
I get this error:
```
ERROR: Received illegal bin number 4294967295 from getBin call.
ERROR: Unable to add record to tree
```
I used grep to search for this number and it's nowhere to be found in either of my files, which makes sense given that my genome isn't that big. So why am I still getting this error?
Thanks for your input!
| The BED files look reasonable.
Maybe there are invisible characters somewhere in the file? Can you try running `mac2unix` or `dos2unix` on them? | biostars | {"uid": 236688, "view_count": 13907, "vote_count": 2} |
Hi all,
I was just wondering if anybody had any experience with coloring something like a UMAP made in ggplot based on the expression of multiple genes at the same time? What I want to do is something like the blend function in Seurat featureplots, but with 3 genes / colors instead of 2.
I'm looking to make something like this:
![Image 1][1]
Where the colors for the genes combine where there is overlap.
What I've gotten to so far is
ggplot(FD, vars = c("UMAP_1", "UMAP_2", "FOSL2", "JUNB", "HES1"), aes(x = UMAP_1, y = UMAP_2, colour = FOSL2)) +
geom_point(size=0.3, alpha=1) +
scale_colour_gradientn(colours = c("lightgrey", colour1), limits = c(0, 0.3), oob = scales::squish) +
new_scale_color() +
geom_point(aes(colour = JUNB), size=0.3, alpha=0.7) +
scale_colour_gradientn(colours = c("lightgrey", colour2), limits = c(0.1, 0.2), oob = scales::squish) +
new_scale_color() +
geom_point(aes(colour = HES1), size=0.3, alpha=0.1) +
scale_colour_gradientn(colours = c("lightgrey", colour3), limits = c(0, 0.3), oob = scales::squish)
Where FD is a data frame containing the information from the seurat object for the UMAP coordinates and the expression levels of the three genes of interest. All I can get is a plot where the points from one layer obscure those below it, I've tried messing around with the colours, gradients, alpha and scales but I'm guessing I'm doing it the wrong way.
![Image 2][2]
If anyone knows of a way to make this work or has any suggestions on something else to try that would be very much appreciated.
[1]: /media/images/e6f45dd8-da0d-4858-950c-74d3f4c4
[2]: /media/images/a661c7cc-7b77-4efe-b921-e1eb4412 | Just in case anyone was searching for an answer for this in the future, I asked the same question over on StackExchange too and was suggested an answer that worked perfectly for me.
https://stackoverflow.com/questions/69609056/colour-umap-based-on-expression-of-multiple-genes-in-ggplot2/ | biostars | {"uid": 9493428, "view_count": 1309, "vote_count": 1} |
So I was struggling with this: Creating a dendrogram with a large dataset (20,000 by 20,000 gene-gene correlation matrix): https://www.biostars.org/p/477332
Now I think I found a good solution, taking a "meaningful" sample of the dataset, and then create a dendrogram-heatmap of the gene-gene correlation matrix generated from the sample.
I have got this far:
cluster3.seurat.obj <- CreateSeuratObject(counts = cluster3.raw.data, project = "cluster3", min.cells = 3, min.features = 200)
cluster3.seurat.obj <- NormalizeData(cluster3.seurat.obj, normalization.method = "LogNormalize", scale.factor = 10000)
cluster3.seurat.obj <- FindVariableFeatures(cluster3.seurat.obj, selection.method = "vst", nfeatures = 2000)
Now I am wondering, how do I extract a data frame or matrix of this Seurat object with the built in function or would I have to do it in a "homemade"-R-way?
I'm hoping it's something as simple as doing this:
cluster3.cells.variable.features <- as.matrix(GetAssayData(cluster3.seurat.obj, slot = "data")[, WhichCells(cluster3.seurat.obj)][, FetchData(cluster3.seurat.obj, var.features)])
I was playing around with it, but couldn't get it...
Any help would be appreciated.
Very Respectfully,
Pratik | You just want a matrix of counts of the variable features?
var_genes <- VariableFeatures(cluster3.seurat.obj)
seurat_df <- GetAssayData(cluster3.seurat.obj)[var_genes,]
| biostars | {"uid": 477549, "view_count": 5955, "vote_count": 1} |
Hello,
I have a problem when using Music. Only missense, nonsense, splicing mutations and indels are included in my data, there are no synonymous mutations, but the tumor do have synonymous mutations.
Is it OK to find SMGs with Music ? Is it OK to define background mutation without silent mutations?
Thank you~ | Read the arguments of `music bmr calc-bmr`, where it defaults to excluding Silent mutations. The documentation is [online here](http://gmt.genome.wustl.edu/packages/genome-music/genome-music-bmr-calc-bmr.html). If you don't want MuSiC to skip Silent mutations, use the argument `--noskip-silent`.
But it is preferable to `--skip-silent` when finding SMGs with somatic mutations, because silent mutations are more likely to be passenger events with lower selective pressure during cancer evolution (though there are exceptions, like when Silent mutations alter promoter motifs). You can try running it with `--skip-silent` and `--noskip-silent`, and compare results. | biostars | {"uid": 109340, "view_count": 1819, "vote_count": 1} |
In trying to solve a problem posed by Stuti Agrawal, I've been trying to make a working example using the Directory type of CWL v1.0 as an input and not getting very far. I tried writing a tool that pics up the files from a Directory and adds them to a zip file. Note, this doesn't add the directory, but rather its contents. So if you have:
/home/foo/one.txt
/home/foo/two.txt
it should add `one.txt` and `two.txt` to the command line, i.e:
zip data.zip one.txt two.txt
I'm not sure if this is the intended use case for Directory? Alternatively, I'd like to able to add `/home/foo` to the command line, yielding:
zip -r data.zip /home/foo
In any event here is a non-working experiment:
#!/usr/bin/env cwl-runner
cwlVersion: v1.0
requirements:
- class: InlineJavascriptRequirement
class: CommandLineTool
baseCommand: ["zip"]
inputs:
testdir:
type: Directory
doc: |
string(s): list files in a directory.
inputBinding:
position: 1
outputs:
zipped_file:
type: File
outputBinding:
glob: data.zip
arguments:
- valueFrom: data.zip
What should be changed to get either my first or my second example command line?
Thanks
| With thanks to Peter's patch to cwltool, here is a working variation:
#!/usr/bin/env cwl-runner
class: CommandLineTool
cwlVersion: v1.0
requirements:
- class: InitialWorkDirRequirement
listing: $(inputs.directory_to_zip.listing)
inputs:
directory_to_zip:
type: Directory
baseCommand: [zip, "--recurse-paths", "-", "."]
outputs:
zipped_file:
type: stdout
format: application/zip
Sample usage with command line input:
$ ls t
bam bam.bai three
$ cwltool zipper.cwl --directory t
/home/michael/cwltool/env/bin/cwltool 1.0.20160820220956
[job zipper.cwl] /tmp/tmpppA9iT$ zip \
--recurse-paths \
- \
. > /tmp/tmpppA9iT/8cccc91d-7bf6-4008-a727-952ad9752114
adding: bam (stored 0%)
adding: three (stored 0%)
adding: bam.bai (stored 0%)
adding: 8cccc91d-7bf6-4008-a727-952ad9752114 (deflated 53%)
Final process status is success
{
"zipped_file": {
"format": "file:///home/michael/cwltool/application/zip",
"checksum": "sha1$4e7aea5191e78a98a66b23cb69652df3a607fbde",
"basename": "8cccc91d-7bf6-4008-a727-952ad9752114",
"location": "file:///home/michael/cwltool/8cccc91d-7bf6-4008-a727-952ad9752114",
"path": "/home/michael/cwltool/8cccc91d-7bf6-4008-a727-952ad9752114",
"class": "File",
"size": 746
}
}
Finally sample usage with an input document
$ cat zipper.in.yml
directory_to_zip:
class: Directory
location: t
$ cwltool zipper.cwl zipper.in.yml
/home/michael/cwltool/env/bin/cwltool 1.0.20160820220956
[job zipper.cwl] /tmp/tmpQqxPjt$ zip \
--recurse-paths \
- \
. > /tmp/tmpQqxPjt/2d0ca78b-6248-46aa-8b1c-0708fd0008a1
adding: bam (stored 0%)
adding: 2d0ca78b-6248-46aa-8b1c-0708fd0008a1 (deflated 11%)
adding: three (stored 0%)
adding: bam.bai (stored 0%)
Final process status is success
{
"zipped_file": {
"format": "file:///home/michael/cwltool/application/zip",
"checksum": "sha1$7c896e9247972af38d3836e621cd9aa66c1b3499",
"basename": "2d0ca78b-6248-46aa-8b1c-0708fd0008a1",
"location": "file:///home/michael/cwltool/2d0ca78b-6248-46aa-8b1c-0708fd0008a1",
"path": "/home/michael/cwltool/2d0ca78b-6248-46aa-8b1c-0708fd0008a1",
"class": "File",
"size": 710
}
}
| biostars | {"uid": 204388, "view_count": 5009, "vote_count": 2} |
I am currently working with a program that generates aritificial FASTQ files when given a reference genome called Artificial FASTQ Generator. [Here][1] is a link to the description of the program and [here][2] is the manual, it says that the program generates paired-end reads (and it does generate two FASTQ files). After aligning the artificial reads (the two FASTQ files generated by Artificial FASTQ Generator) to the reference genome as paired-end reads using Bowtie 2 I got the following result:
```
892589 (100.00%) were paired; of these:
892585 (100.00%) aligned concordantly 0 times
4 (0.00%) aligned concordantly exactly 1 time
0 (0.00%) aligned concordantly >1 times
----
892585 pairs aligned concordantly 0 times; of these:
870486 (97.52%) aligned discordantly 1 time
----
22099 pairs aligned 0 times concordantly or discordantly; of these:
44198 mates make up the pairs; of these:
7154 (16.19%) aligned 0 times
15716 (35.56%) aligned exactly 1 time
21328 (48.26%) aligned >1 times
99.60% overall alignment rate
```
After aligning both FASTQ files as single end reads, I got the following:
```
1785178 (100.00%) were unpaired; of these:
7154 (0.40%) aligned 0 times
1755694 (98.35%) aligned exactly 1 time
22330 (1.25%) aligned >1 times
99.60% overall alignment rate
```
What I do not understand is why these reads are aligning as single-end reads and not as paired-end reads as expected? Is anybody familiar with both programs that can help explain this?
EDIT:
Here are the outputs:
- [1st FASTQ output of AFG][3]
- [2nd FASTQ output of AFG][4]
- [SAM output of Bowtie 2][5] (single-end alignment)
EDIT 2:
Here is the output of the paired-end alignment.
[SAM output of Bowtie 2][6] (paired-end alignment)
[1]: http://sourceforge.net/projects/artfastqgen/
[2]: http://sourceforge.net/p/artfastqgen/wiki/Home/
[3]: https://drive.google.com/file/d/0B3xfexs8wgRTS2tiYkROanJYdnM/edit?usp=sharing
[4]: https://drive.google.com/file/d/0B3xfexs8wgRTRWdxeV9iZkhoOGc/edit?usp=sharing
[5]: https://drive.google.com/file/d/0B3xfexs8wgRTOVV2Rl9KSUxSc3c/edit?usp=sharing
[6]: https://drive.google.com/file/d/0B3xfexs8wgRTR1NDM3A3TE5JX2c/edit?usp=sharing | Maybe you should post an example of the output, a few read pairs should be sufficient.
There could be many reasons, e.g.:
- Bug or wrong use of the simulator: mate pairs not on complementary strands
- too large insert size
- Bug: annotated pairs do not pair
- too low simulated quality scores (single-end do align though, so not in this case)
- too high simulated error rate (dto.)
Maybe you should use a simulator that is better tested, see:
https://www.biostars.org/p/2194/ | biostars | {"uid": 104086, "view_count": 3220, "vote_count": 2} |
All proteins on UniProt have a unique accession number. Ex "O15169" is the accession for human Axin 1.
Other RDF stores referring to proteins on UniProt use this accession (eg [Pathway Commons reference][2])
[This document][1] describes the RDF schema for UniProt.
Where is the UniProt accession in this RDF schema?
[1]: https://www.uniprot.org/core/
[2]: http://rdf.pathwaycommons.org/describe/?url=http://pathwaycommons.org/pc12/UnificationXref_uniprot_knowledgebase_O15169 | In the UniProt RDF model, the accession is only in the IRI of the form `http://purl.uniprot.org/uniprot/${ACCESSION}`.
To go from an accession string in pathway commons to a IRI one uses a SPARQL snippet like:
VALUES ?acc { "P05067" }
BIND(IRI(CONCAT("http://purl.uniprot.org/uniprot", ?acc)) AS ?entry)
There are two reasons that we don't have the primary accession as a string in our RDF or SPARQL endpoint.
1. Avoiding false joins, an UniProt accession. Might also be used to identify something completely else, without the IRI part false joins can lead to wrong results.
2. Adding a string for each identifier adds hundreds of millions of extra triples and strings in the database which will negatively impact performance and storage. | biostars | {"uid": 462218, "view_count": 810, "vote_count": 1} |
I have a list of accession numbers, how can I search for there function in Pfam db http://pfam.xfam.org/ as a bulk?
For example:
If I have this accession **PF07714.12** and I searched manually I will get;
> A tyrosine kinase is an [enzyme][1] that can transfer a [phosphate][2] group from [ATP][3] to a [protein][4] in a cell. It functions as an "on" or "off" switch in many cellular functions. Tyrosine [kinases][5] are a subclass of [protein kinase][6].
>
> The phosphate group is attached to the [amino acid][7] [tyrosine][8] on the protein. Tyrosine kinases are a subgroup of the larger class of [protein kinases][9] that attach phosphate groups to other amino acids ([serine and threonine][10]). [Phosphorylation][11] of proteins by kinases is an important mechanism in communicating signals within a cell ([signal transduction][12]) and regulating cellular activity, such as cell division.
>
> Protein kinases can become mutated, stuck in the "on" position, and cause unregulated growth of the cell, which is a necessary step for the development of cancer. Therefore, kinase inhibitors, such as[imatinib][13], are often effective cancer treatments.
How can I do it for a list of accessions?
[1]: http://en.wikipedia.org/wiki/Enzyme
[2]: http://en.wikipedia.org/wiki/Phosphate
[3]: http://en.wikipedia.org/wiki/Adenosine_triphosphate
[4]: http://en.wikipedia.org/wiki/Protein
[5]: http://en.wikipedia.org/wiki/Kinases
[6]: http://en.wikipedia.org/wiki/Protein_kinase
[7]: http://en.wikipedia.org/wiki/Amino_acid
[8]: http://en.wikipedia.org/wiki/Tyrosine
[9]: http://en.wikipedia.org/wiki/Protein_kinase
[10]: http://en.wikipedia.org/wiki/Serine/threonine-specific_protein_kinase
[11]: http://en.wikipedia.org/wiki/Phosphorylation
[12]: http://en.wikipedia.org/wiki/Signal_transduction
[13]: http://en.wikipedia.org/wiki/Imatinib | Unlike what you have shown here, if **one sentence description** is enough to describe about the family you can use `hmmfetch` which first extract profiles and from that file you can separate ACC & DESC lines easily, ACC - accession id, DESC - description about family. [Here][1] you can download all pfam family HMM profiles in one file, then use it as a source to extract families of your interest.
[1]: ftp://ftp.ebi.ac.uk/pub/databases/Pfam/current_release | biostars | {"uid": 135092, "view_count": 4380, "vote_count": 1} |
Hello,
I want to extract some specific strings in csv file, using either excel, R or Python.
for example as below: I want to find string from column A in column B and return in column C with 5 amino acid before and after **N**; thanks!!
```
A B C
INETTDFR MHRFLLMLLFPFSDNRPMMFFRSFIVFFFLIFFASNVSSRKQTYVIHT IVGKINETTDF
VTTSTKHIVTSLFNSLQTENINDDDFSLPEIHYIYENAMSGFSATLTDDQLDT
VKNTKGFISAYPDELLSLHTTYSHEFLGLEFGIGLWNETSLSSDVIIGLVDTG
ISPEHVSFRDTHMTPVPSRWRGSCDEGTNFSSSECNKKIIGASAFYKGYE
SIVGKINETTDFRSTRDAQGHGTHTASTAAGDIVPKANYFGQAKGLASGM
RFTSRIAAYKACWALGCASTDVIAAIDRAILDGVDVISLSLGGSSRPFYVDP
IAIAGFGAMQKNIFVSCSAGNSGPTASTVSNGAPWLMTVAASYTDRTFPAIV
RIGNRKSLVGSSLYKGKSLKNLPLAFNRTAGEESGAVFCIRDSLKRELVEGK
IVICLRGASGRTAKGEEVKRSGGAAMLLVSTEAEGEELLADPHVLPAVSLGF
SDGKTLLNYLAGAANATASVRFRGTAYGATAPMVAAFSSRGPSVAGPEIAKP
DIAAPGLNILAGWSPFSSPSLLRSDPRRVQFNIISGTSMACPHISGIAALIKSV
HGDWSPAMIKSAIMTTARITDNRNRPIGDRGAAGAESAATAFAFGAGNVDPT
RAVDPGLVYDTSTVDYLNYLCSLNYTSERILLFSGTNYTCASNAVVLSPGDLN
YPSFAVNLVNGANLKTVRYKRTVTNVGSPTCEYMVHVEEPKGVKVRVEPKVL
KFQKARERLSYTVTYDAEASRNSSSSSFGVLVWICDKYNVRSPIAVTWE
``` | Using Excel*:
=MID(B1,FIND(A1,B1)+FIND("N",A1)-6,5) & "N" & MID(B1,FIND(A1,B1)+FIND("N",A1),5)
*Don't use Excel :) | biostars | {"uid": 109077, "view_count": 4342, "vote_count": 1} |
Hey everyone,
I'm inexperienced with statistics, and want to perform a regression between two diseases. I would really appreciate some clarification if my understanding of when to include an interaction term is correct.
Let's call the diseases disease Y and disease X. I know that disease Y is age-dependent, i.e. it's more likely to be encountered in older individuals and it becomes progressively worse with age. So I think the regression should definitely include age as a covariate:
Y ~ age + X
I'm not sure whether disease X is also age-dependent, but it might be. My planned approach was to look into the data, check if an independent student's t-test detects a significant difference in the age distributions between people with / without disease X. If yes, I would correct the regression formula to include an interaction term:
Y ~ age + age:X + X
Would this approach be correct?
Additionally, would it matter if my variable Y represents case/control status or disease severity (i.e. logistic vs linear regression)?
| It's best practice to first check if your variables are correlated. If they are, you should either drop one or combine them into one variable.
In R:
cor.test(your_data$age, your_data$X)
I would drop one of the variables if r >= 0.5, although others may use a different cutoff. If they are correlated, I would keep the variable with the lowest p-value. Alternatively, you could combine age and X into one variable by adding them or taking their average.
To find p-values:
model = lm(Y ~ age + X, data = your_data)
summary(model)
If age and X are not correlated, then you can see if there is an interaction.
int.model = lm(Y ~ age + X + age:X, data = your_data)
summary(int.model)
If the interaction term has a significant p-value, then you'll want to include it in your model. If not, then you'll want to drop it.
You can use either linear or logistic regression. For logistic regression, you would use the following:
logit.model = glm(Y ~ age + X + age:X, data = your_data, family = binomial)
summary(logit.model) | biostars | {"uid": 9534805, "view_count": 646, "vote_count": 1} |
<p>Do you know any <strong>public</strong> scientific SQL server ?</p>
<p>for example, I would cite:</p>
<ul>
<li>UCSC <a href='http://genome.ucsc.edu/FAQ/FAQdownloads#download29'>http://genome.ucsc.edu/FAQ/FAQdownloads#download29</a></li>
<li>ENSEMBL <a href='http://uswest.ensembl.org/info/data/mysql.html'>http://uswest.ensembl.org/info/data/mysql.html</a></li>
<li>GO <a href='http://www.geneontology.org/GO.database.shtml#mirrors'>http://www.geneontology.org/GO.database.shtml#mirrors</a></li>
</ul>
<p>(I'll give a +1 to each correct answer)</p>
| This is quite an important one for people doing mouse work, though it is important to note that JAX offer Mart and Batch-Query functionality through the web site as well which may well suit many peoples needs.
- Direct SQL Access to MGI
- <a href="http://www.informatics.jax.org/software.shtml#sql">http://www.informatics.jax.org/software.shtml#sql</a>
Note that this is a public 'free' service, but that you do need to contact user support to get your login and password. They are also happy to provide some custom SQL scripts to get you started. | biostars | {"uid": 474, "view_count": 11197, "vote_count": 22} |
Heys,
I'm working with bedtools trying to find out the coverage of several target regions.
I have my bam files and my target regions in a .bed file and I'm running this:
bedtools bamtobed -i WA01.bam | bedtools coverage -a - -b mit.bed > exons.mit.coverage
When I open the exons.mit.coverage file, in the coverage section I just have 0s in the areas that are not matching my target genes and 1s in the areas of my target genes, as I have **coverage per read**.
My question: How can I obtain coverage per gene and not coverage per read?
I imagine I can filter this out, selecting the reads with a good quality score per gene, but I think this should be already implemented anywhere else that I don't know
Thanks for your help! :) | Switch `-a` and `-b`.
bedtools bamtobed -i WA01.bam | bedtools coverage -b - -a mit.bed > exons.mit.coverage | biostars | {"uid": 433473, "view_count": 1702, "vote_count": 1} |
According to http://www.ebi.ac.uk/~zerbino/velvet/, velvet can be multithreaded. But how to do it? When I do
```
./velveth
Usage:
./velveth directory hash_length {[-file_format][-read_type][-separate|-interleaved] filename1 [filename2 ...]} {...} [options]
```
My question is how to run velveth multithreaded? Where is the multi-thread flag (usually `-t` or `-p`)? I'm running 1.2.10. | <p>For threading to work, Velvet has to be installed with <code>make 'OPENMP=1'</code>. Then you can control threading via <code>export OMP_NUM_THREADS=7</code>. See <a href="https://helix.nih.gov/Applications/velvet_manual.pdf">Velvet Manual</a> Section 2.3.6.</p>
| biostars | {"uid": 142938, "view_count": 5429, "vote_count": 1} |
As per the title: why?
Does the preliminary PCA step merely maximise the chances of identifying the clusters that tSNE / UMAP later identify?
When doing this, in tutorials, it seems that people blindly choose a number of PCs as input to tSNE / UMAP, without checking the amount of variation explained by the PCs. For example, 20 PCs may only account for 30% overall variation.
Assuming one follows a standard procedure for, e.g., scRNA-seq, is this one step too many. A pipeline could be:
1. normalisation of raw counts
2. transformation of normalised counts
3. PCA-transformation (may include an additional pre-scaling step)
4. tSNE/UMAP
Kevin | At least from a clustering perspective, I'd probably try it both ways to be on the safe side, i.e. with PCA to get the top N PCs and without. I'm a bit skeptical of reducing to N PCs for clustering because there is inevitable information loss. The same will apply for t-SNE, UMAP, etc. I'd prefer to use the most variable genes instead.
Although I think it is less of a problem when just trying to visualise the data, rather than define some new cell types using cluster analysis where we might want to be a bit more cautious. | biostars | {"uid": 381993, "view_count": 8650, "vote_count": 7} |
Hello,
I have multiple fasta sequences that are like this:
>2p__scaffold_2__5799__6580__-__778568__0.00__0.00
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAACTGAGAGTCGGCGATGAGCAGCCGGA
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAA
>2p__scaffold_2__5799__6580__+__778569__0.00__0.00
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAACTGAGAGTCGGCGATGAGC
>1p__scaffold_2__11235__11438__-__830827__0.00__0.00
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAACTGAGAGTCGGCGATGAGCAGCCGGA
GCTTCAATCCAGGGGATCGAGGAGATCCAAAGCAGCAGAAGCGGCTCGACGATGGTGAGG
ATTCGGGATCGGATTCAGCGCTCGTCGGGACTGG
>1p__scaffold_2__33129__34129__+__811706__0.00__0.00
GCTGGCGACGGATCTA
And I want to keep just the "> + ID" (numbers after `__+/-__` and before __0.00_0.00)
So I expect an output like this:
>778568
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAACTGAGAGTCGGCGATGAGCAGCCGGA
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAA
>778569
GCTGGCGACGGATCTAGGCTCAGCGCAGAAGCAACTGAGAGTCGGCGATGAGC
I searched for it and tried this:
sed 's@.*__-__@@' input.fa > output.fa
That removed `__-__` and everything before it, including the ">" that I wanted to keep.
I also tried this to remove everything between ">" and `__-__`
sed -e 's/\>//' -e 's/\__-__.*//' input.fa > output.fa
But this removed everything after `__-__`
And this, that removed __0.00_0.00
sed 's/__0.00.*$//' input.fa > output.fa
Thank you for your help. | This should work if your headers follow the pattern that you specified:
sed 's/[>_]\+/_/g' yourfile.fasta | cut -f 8 -d _ | sed 's/^\([0-9]\)/>\1/'
| biostars | {"uid": 302838, "view_count": 1721, "vote_count": 1} |
Hello,
I want to extract some specific strings in csv file, using either excel, R or Python.
for example as below: I want to find string from column A in column B and return in column C with 5 amino acid before and after **N**; thanks!!
```
A B C
INETTDFR MHRFLLMLLFPFSDNRPMMFFRSFIVFFFLIFFASNVSSRKQTYVIHT IVGKINETTDF
VTTSTKHIVTSLFNSLQTENINDDDFSLPEIHYIYENAMSGFSATLTDDQLDT
VKNTKGFISAYPDELLSLHTTYSHEFLGLEFGIGLWNETSLSSDVIIGLVDTG
ISPEHVSFRDTHMTPVPSRWRGSCDEGTNFSSSECNKKIIGASAFYKGYE
SIVGKINETTDFRSTRDAQGHGTHTASTAAGDIVPKANYFGQAKGLASGM
RFTSRIAAYKACWALGCASTDVIAAIDRAILDGVDVISLSLGGSSRPFYVDP
IAIAGFGAMQKNIFVSCSAGNSGPTASTVSNGAPWLMTVAASYTDRTFPAIV
RIGNRKSLVGSSLYKGKSLKNLPLAFNRTAGEESGAVFCIRDSLKRELVEGK
IVICLRGASGRTAKGEEVKRSGGAAMLLVSTEAEGEELLADPHVLPAVSLGF
SDGKTLLNYLAGAANATASVRFRGTAYGATAPMVAAFSSRGPSVAGPEIAKP
DIAAPGLNILAGWSPFSSPSLLRSDPRRVQFNIISGTSMACPHISGIAALIKSV
HGDWSPAMIKSAIMTTARITDNRNRPIGDRGAAGAESAATAFAFGAGNVDPT
RAVDPGLVYDTSTVDYLNYLCSLNYTSERILLFSGTNYTCASNAVVLSPGDLN
YPSFAVNLVNGANLKTVRYKRTVTNVGSPTCEYMVHVEEPKGVKVRVEPKVL
KFQKARERLSYTVTYDAEASRNSSSSSFGVLVWICDKYNVRSPIAVTWE
``` | Something like this should get you started. I haven't tested this, though, and you may need to change `delimiter` and `quotechar` parameters based on what your input and output look like:
```
#!/usr/bin/env python
import csv
import sys
padding = 5
assert padding >= 0, 'padding is less than zero, which does not make sense'
residues_of_interest = 'N'
roi_length = len(residues_of_interest)
csv_writer = csv.writer(sys.stdout, delimiter=',', quotechar='\"', quoting=csv.QUOTE_ALL)
for row in csv.reader(iter(sys.stdin.readline, ''), delimiter=',', quotechar='\"'):
A = row[0]
B = row[1]
pos_roi_in_A = A.find(residues_of_interest)
if pos_roi_in_A == -1:
continue # skip over lines where residues-of-interest string is not in A
pos = B.find(A) + pos_roi_in_A # anchor 'pos' value on the start position of ROI
lower_bound = padding
upper_bound = len(B) - padding
#
# consider four cases of a bounded string search result
#
if pos == -1:
C = None
elif pos < lower_bound:
C = B[0:(pos + roi_length + padding)]
elif pos > upper_bound:
C = B[(pos - padding):upper_bound]
else:
C = B[(pos - padding):(pos + roi_length + padding)]
#
# write A, B and C to standard output
#
csv_writer.writerow([A, B, C])
```
You might run it like so:
$ padded_substring_finder.py < in.csv > out.csv | biostars | {"uid": 109077, "view_count": 4342, "vote_count": 1} |
hi friends,
i RNA-seq protocol i read...
tophat -p 8 -G genes.gtf -o C1_R1_thout genome C1_R1_1.fq C1_R1_2.fq
by which i have accepted_hits.bam
cufflinks -p 8 -o C1_R1_clout C1_R1_thout/accepted_hits.bam
then saying that :
> Create a file called assemblies.txt that lists the assembly file for each sample. The file should contain the following lines:
./C1_R1_clout/transcripts.gtf
./C2_R2_clout/transcripts.gtf
./C1_R2_clout/transcripts.gtf
./C2_R1_clout/transcripts.gtf
./C1_R3_clout/transcripts.gtf
./C2_R3_clout/transcripts.gtf
then this command
cuffmerge -g genes.gtf -s genome.fa -p 8 assemblies.txt
what is the `assemblies.txt` please??? how i can create such a file by *cufflinks output* that as i saw is a file named `QC_filtered.fastq`
thanks for your simpathy | <p>The output of cufflinks is<code> transcripts.gtf</code> . If you have many samples, cufflinks creates transcripts.gtf ( i.e assembles transcripts) file for each samples and these needs to be merged by cuffmerge,</p>
| biostars | {"uid": 156992, "view_count": 9460, "vote_count": 3} |
Hi all,
I need to assign GO terms to DE genes in R. `ClusterProfiler` seems to be a popular package for that.
But for some reason it either fails to detect GO terms or its GO terms do not coincide with other tools.
library(clusterProfiler)
library(org.Hs.eg.db)
###mtDNA genes
genes<-c("ENSG00000198886","ENSG00000198888","ENSG00000198763","ENSG00000198840","ENSG00000212907","ENSG00000198786","ENSG00000198695","ENSG00000198727","ENSG00000198804","ENSG00000198712","ENSG00000198938","ENSG00000198899","ENSG00000228253")
ego<-enrichGO(genes, OrgDb = org.Hs.eg.db, keytype ="ENSEMBL",ont = "CC",pAdjustMethod = "fdr",pvalueCutoff = 0.1, qvalueCutoff = 0.5)
dotplot(ego, showCategory=30)
The same genes with `Panther` as expected show mtDNA related GO terms. Other genes from my dataset get enrichments, so apparently the problem is not in gene IDs.
What am I doing wrong?
Cheers,
| With the hint from @Guangchuang in the post above, I resolved the problem by updating `clusteProfiler` to the latest version.
For that I had to update R to v. 3.5 for my Ubuntu 16.04 https://www.r-bloggers.com/updating-r-on-ubuntu/
Then install the latest bioconductor version https://www.bioconductor.org/install/
And resolve the igragh fortrat4 dependancy issue https://ashokragavendran.wordpress.com/2017/10/24/error-installing-rigraph-unable-to-load-shared-object-igraph-so-libgfortran-so-4-cannot-open-shared-object-file-no-such-file-or-directory/
Hope this will save several hours for people with the same problem
| biostars | {"uid": 371324, "view_count": 3065, "vote_count": 1} |
Hi, I'm pretty new to the scRNA-seq world and while working on my own sets of data, I'm starting to wonder when should the batch correction algorithm be used appropriately.
Let's say we have a Day0, a Day1, a Day2 and a Day3 scRNA-seq sample.
To elaborate, starting from Day0, assume we treated certain chemical, and sampled it on a daily basis during the course of experiment.
**Would it be ok or reasonable to apply batch correction algorithm (e.g. CCA) to this aggregation of samples?
I mean, is CCA algorithm designed for this kind of experiment design?**
From the experiment from [Kang et al., 2017][1] which is comprised of PBMC, splitted into a control group and a stimulated group treated with interferon beta, [they state that][2] "the repsonse to interferon caused cell type specific gene expression changes that makes a joint analysis of all the data difficult with cells clustering both by stimulation condition and by cell type". But is it reasonable?
My understanding is that if you are to use batch correction you should have biological or technical batches from the "same condition". So if you have replicate samples with the same condition and when somehow they are separated from each other for technical reason, it's appropriate to use batch correction.
Going back to the supposed experiment I stated above, I think (maybe i'm wrong and i am most of time) it's not reasonable to apply batch correction to this Day0-4 experiment.
Can someone give me some clear explanation to the use of batch correction?
Thank you.
Ryan
[1]: https://www.nature.com/articles/nbt.4042
[2]: https://satijalab.org/seurat/v3.0/immune_alignment.html | > if you are to use batch correction you should have biological or technical batches from the "same condition"
From the [integration vignette][1]: "These methods aim to identify shared cell states that are present across different datasets, even if they were collected from different individuals, **experimental conditions**, technologies, or even species"
Thus, the "official" answer is that different conditions are fine.
Really, it depends on the questions you want to ask and on the data that you have. For example, if all your time points segregate and form distinct clusters, it's going to be hard to present any kind of coherent analysis.
[1]: https://satijalab.org/seurat/v3.0/integration.html | biostars | {"uid": 458870, "view_count": 1333, "vote_count": 1} |
Dear all, I have easy script in bash using awk language. I filtrate bam file by GC content (see at code), but I need to get on output bam file too with header. Could you help me with this? thank you My code:
#!/bin/bash
for i in 54321*.bam
do
samtools view -h /home/filip/Desktop/Analyza\ NIFTY\ and\ CNVs/NIFTY\ pooling009/$i | awk '{ n=length($10); print gsub(/[GCCgcs]/,"",$10)/n,"\t",$0 }' | awk '($1 <= 0.6 && $1 >= 0.3){print $0}' | awk '!($1="")' | samtools -bS - > z$i;
done | you could use your code without including the header in a first samtools run (and you wouldn't have to check whether each line is a header or a read, you'll only be dealing with reads), and then embed the header from the original bam in a subsequent samtools run. something like this:
for bamfile in *bam; do
samtools view -H $bamfile > temp.sam
samtools view $bamfile | yourcode >> temp.sam
samtools view -bS temp.sam > mod_$bamfile
done | biostars | {"uid": 102752, "view_count": 4435, "vote_count": 2} |
hello all,
i am having a hard time in using bcftools set set some variants in my vcf to missing.
as i understand missing2ref is no longer used and replaced with setGT. having said that, it is not clear how to use it.
i need to set any variant with genotype quality (GQ) < 20 to missing. this will require that i use the plugin +fill-tags at the first stage, but then how do i use the setGT plugin? how is the filter command written?
i am so lost here. thanks in advance | Below is a Python API solution using the `pyvcf` [submodule](https://sbslee-fuc.readthedocs.io/en/latest/api.html?highlight=VcfFrame.from_dict#module-fuc.api.pyvcf) from the `fuc` [package](https://sbslee-fuc.readthedocs.io/en/latest/readme.html) I wrote.
Imagine you have the following data:
```
>>> from fuc import pyvcf
>>> data = {
... 'CHROM': ['chr1', 'chr1', 'chr1', 'chr1'],
... 'POS': [100, 101, 102, 103],
... 'ID': ['.', '.', '.', '.'],
... 'REF': ['G', 'T', 'A', 'A'],
... 'ALT': ['A', 'C', 'T', 'C'],
... 'QUAL': ['.', '.', '.', '.'],
... 'FILTER': ['.', '.', '.', '.'],
... 'INFO': ['.', '.', '.', '.'],
... 'FORMAT': ['GT:GQ:DP', 'GT:GQ:DP', 'GT:GQ:DP', 'GT'],
... 'A': ['0/0:48:3', '0/0:19:3', './.:.:.', '0/1'],
... 'B': ['0/1:3:5', '0/1:20:8', '0/0:29:12', '0/0'],
... 'C': ['0/0:16:3', '1/0:32:8', '0/1:25:9', '0/0'],
... }
>>> vf = pyvcf.VcfFrame.from_dict([], data)
>>> vf.df
CHROM POS ID REF ALT QUAL FILTER INFO FORMAT A B C
0 chr1 100 . G A . . . GT:GQ:DP 0/0:48:3 0/1:3:5 0/0:16:3
1 chr1 101 . T C . . . GT:GQ:DP 0/0:19:3 0/1:20:8 1/0:32:8
2 chr1 102 . A T . . . GT:GQ:DP ./.:.:. 0/0:29:12 0/1:25:9
3 chr1 103 . A C . . . GT 0/1 0/0 0/0
```
We can then define a custom method (`one_row`) to be applied to each row to convert genotypes calls with GQ < 20 to missing:
```
>>> def one_row(r):
... format_list = r.FORMAT.split(':')
... # This method automatically finds the appropriate missing value ('./.', '.', './.:.:.', etc.).
... missval = pyvcf.row_missval(r)
...
... if 'GQ' in format_list:
... i = format_list.index('GQ')
... else:
... # This row doesn't have GQ, return the entire row as is.
... return r
...
... def one_gt(g):
... gq = g.split(':')[i]
...
... if gq.isnumeric():
... gq = int(gq)
... else:
... # This genotype doesn't have a numeric GQ value (probably has missing value '.'), keep it as is.
... return g
...
... if gq >= 20:
... # This genotype has GQ >= 20, keep it as is.
... return g
... else:
... return missval
...
... r[9:] = r[9:].apply(one_gt)
...
... return r
...
```
We then apply the method:
```
>>> vf.df = vf.df.apply(one_row, axis=1)
>>> vf.df
CHROM POS ID REF ALT QUAL FILTER INFO FORMAT A B C
0 chr1 100 . G A . . . GT:GQ:DP 0/0:48:3 ./.:.:. ./.:.:.
1 chr1 101 . T C . . . GT:GQ:DP ./.:.:. 0/1:20:8 1/0:32:8
2 chr1 102 . A T . . . GT:GQ:DP ./.:.:. 0/0:29:12 0/1:25:9
3 chr1 103 . A C . . . GT 0/1 0/0 0/0
```
Note that you can easily read and write VCF files too:
```
vf = pyvcf.VcfFrame.from_file('in.vcf')
vf.to_file('out.vcf')
```
Let me know in the comment if you have any questions. | biostars | {"uid": 9499071, "view_count": 1357, "vote_count": 1} |
Hello,
I've my BLAST alignment as below:
Query 1 AGTAAAGCCGACTCGGCTATCCATGGGTGAGAACCTAAAGCCGAGTCGGCTTTAAGTTCT 60
|||||||||||||||||| |||||||||| ||||||||||||||||||||||||||||||
Sbjct 2913022 AGTAAAGCCGACTCGGCTGTCCATGGGTGGGAACCTAAAGCCGAGTCGGCTTTAAGTTCT 2913081
Query 61 GGAAAGTCCCATTTGTCCAGCAGGAAAAGCCGACTCGGCTTTCCTGGTGTTGGGGCAAAA 120
||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||
Sbjct 2913082 AGAAAGTCCCATTTGTCCAGCAGGAAAAGCCGACTCCGCTTTCCTGGTGTTGGGGCAAAA 2913141
Query 121 GCCGACTCGGCTTTTTCCTCTGTTATGAGC**R**TTGGtttttttCCCGTTTTCTTTGAGTAA 180
||||||||||| |||||||||||||||||| ||||| ||||| |||||||||||||||||
Sbjct 2913142 GCCGACTCGGCCTTTTCCTCTGTTATGAGC**G**TTGGTCTTTTTTCCGTTTTCTTTGAGTAA 2913201
Query 181 TTGCTTTGGATTCTTTCACTTACGGTTCTTGATTTGTAGAGTTATAAGGGAGTATTAAGG 240
|||||||||||||||||||||| || ||||||||||| ||||||||||||||||||||||
Sbjct 2913202 TTGCTTTGGATTCTTTCACTTATGGCTCTTGATTTGTGGAGTTATAAGGGAGTATTAAGG 2913261
Query 241 AGAATAATACTCATGAATGGCGTTGAATTGGATGATCATCAATATGATCATTAAGAGTGA 300
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 2913262 AGAATAATACTCATGAATGGCGTTGAATTGGATGATCATCAATATGATCATTAAGAGTGA 2913321
Query 301 T 301
|
Sbjct 2913322 T 2913322
I want the nucleotide thats exactly aligning with the letter R in the query sequence. As you can see the R aligns with a G in the database.
Is there a tool in Biopython or R that can help me code and get me the output?
I've 26101 such sequences where I need the nucleotide that matches with the letters W,R,K,Y,M,N,S in the query sequence. Manually browsing through the alignment and deciding the nucleotide thats matching with it will not cut it as you can see.
Any help would be appreciated. Thanks! | Not the most beautiful solution..
awk '{if(/^Q/ || /^S/){print $3}}' input.file \
| paste - - \
| awk 'BEGIN{OFS=FS="\t"}{for(i=1;i<length($1);i++){L=substr($1,i,1); if(L!="A" && L!="T" && L!="G" && L!="C" && L!="a" && L!="t" && L!="g" && L!="c"){print substr($1,i,1),substr($2,i,1)}}}'
Output:
* *
* *
R G
* *
* *
| biostars | {"uid": 366298, "view_count": 873, "vote_count": 1} |
<p>Hello,</p>
<p>I am developing customized pipelines for ChIP-seq analysis using Snakemake. I want share it, so I created model workflows that people can execute immediatly after downloading the code. It handles file conversion, mapping, peak-calling... And uses public data from GEO database. However it requires people to download these data themselves. I would like to include an automatic download of the data (sra or fastq files), ideally by using GSM/GSE or SRR identifiers.</p>
<p>So far I've found several ways:</p>
<p>* SRA toolkit's fastq-dump function.</p>
<pre>
fastq-dump --outdir <outdir> <srr_ids></pre>
<p>However this way is insanely slow (as stated <a href="https://www.biostars.org/p/91885/">here).</a></p>
<p>* SRAdb R package</p>
<pre>
getSRAfile( in_acc = "<srr_ids>", sra_con = sra_con, destDir = <dir>, fileType = 'sra' )</pre>
<p>This requires using this command first:</p>
<pre>
geometadbfile <- getSRAdbFile(destdir = <dir>, destfile = "SRAmetadb.sqlite.gz")</pre>
<p>which downloads locally an sqlite file of 16Go. Could be fine if I were to use it locally, but I don't want users of my pipeline to be forced to do so...</p>
<p>* Biopython's Bio.Geo module</p>
<p>Not sure how this one works... <a href="http://biopython.org/DIST/docs/tutorial/Tutorial.html#htoc123">http://biopython.org/DIST/docs/tutorial/Tutorial.html#htoc123</a></p>
<p>The object Entrez.esearch doesn't help me finding out the ftp URL or so.<br />
<br />
<br />
I think there should be a way to download data in a more simple way?</p>
<p>Any idea will be greatly appreciated!</p>
| I just found out <a href="https://www.biostars.org/p/111040/">here</a> that I can download the sra files with SRA toolkit, using the prefetch command:
prefetch <SRR ID>
It's quite fast, the only issue is that it seems there is no output directory option. It's worth mentioning that the data is automatically downloaded to `/home/<USER>/ncbi/public/sra/<SRR ID>.sra` (not mentioned in the doc!)
I can then run fastq-dump to get fastq files:
fastq-dump --outdir <output directory> /home/<USER>/ncbi/public/sra/<SRR ID>.sra
Surprisingly, it looks a lot faster than doing these 2 steps at once with fast-dump... | biostars | {"uid": 172009, "view_count": 2913, "vote_count": 1} |
<p>hello friends,</p>
<p>i have a list of GO terms, for example GO:0005975, how i can fine that which of these GO terms is related with cirus fruit senescence please???</p>
| Try [DAVID][1] for the start, than you should read the manuals of some Bioconductor packages.
- [topGO][2]
- [GOexpress][3]
- https://www.biostars.org/p/245/
- https://www.biostars.org/p/8190/
- https://www.biostars.org/p/85514/
- [GO Analysis][4]
- [GO analysis with R code][5]
Read properly and perform the commands for the dataset provided by the packages. Don't simply try to copy paste, to do analysis yourself. Understand the commands properly, what's going on and then try to implement the codes accordingly.
[1]: http://david.abcc.ncifcrf.gov/
[2]: http://www.bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf
[3]: http://www.bioconductor.org/packages/release/bioc/vignettes/GOexpress/inst/doc/GOexpress-UsersGuide.pdf
[4]: http://cals.arizona.edu/~anling/MCB516/lecture22.pdf
[5]: http://davetang.org/muse/2010/11/10/gene-ontology-enrichment-analysis/ | biostars | {"uid": 150787, "view_count": 2519, "vote_count": 1} |
Hello,
I'd like to filter for sites that have a DP>5 in my normal sample and DP>10 in the tumor. My vcf has two samples columns, TUMOR NORMAL. How do I do that with a bcftools expression? I see in the documentation how to apply the filter for one or all samples in a vcf row but I couldn't find a way to specify the sample name.
Thanks a lot. | Actually bcftools supports this also. This worked for me:
`bcftools filter -s MY_FILTER -m + -e "FMT/DP[0] <= 5 || FMT/DP[1] <= 10" my.vcf`
which appends MY_FILTER based on the exclusion criteria | biostars | {"uid": 187558, "view_count": 4260, "vote_count": 2} |
I've got a list of codding genes and I would like to get the cellular location of the expressed proteins, all at once. I already looked up on uniprot but wasn't able to solve it. | You could use data from the [human protein atlas][1] or [neXtProt][2] for this.
[1]: http://www.proteinatlas.org/humanproteome
[2]: http://www.nextprot.org/ | biostars | {"uid": 154549, "view_count": 2652, "vote_count": 1} |
I am trying to run a basic Tophat2 command but it goes wrong somewhere. Perhaps anyone has experience with this error message? I'm running Tophat2 within a Virtual Box with BioLinux, writing output to shared directories with the windows computer. When specifying output directory to a non-shared directory the same error occurs.
**Command**:
tophat2 \
-o /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq/ \
--keep-tmp \
-p 8 \
/home/koen/Host/Stage_Enschede/methods_Bowtie2/indexes_chromFA/genome /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq/3_S3_L001_R1_001_accepted.fq
**Error**:
[FAILED] Error running /usr/bin/tophat_reports
**Log**:
[2014-06-02 13:37:09] Beginning TopHat run (v2.0.9)
-----------------------------------------------
[2014-06-02 13:37:09] Checking for Bowtie
Bowtie version: 2.1.0.0
[2014-06-02 13:37:09] Checking for Samtools
Samtools version: 0.1.19.0
[2014-06-02 13:37:09] Checking for Bowtie index files (genome)..
[2014-06-02 13:37:09] Checking for reference FASTA file
[2014-06-02 13:37:09] Generating SAM header for /home/koen/Host/Stage_Enschede/methods_Bowtie2/indexes_chromFA/genome
format: fastq
quality scale: phred33 (default)
[2014-06-02 13:38:08] Preparing reads
left reads: min. length=242, max. length=300, 541 kept reads (0 discarded)
[2014-06-02 13:38:08] Mapping left_kept_reads to genome genome with Bowtie2
[2014-06-02 13:38:39] Mapping left_kept_reads_seg1 to genome genome with Bowtie2 (1/12)
[2014-06-02 13:39:09] Mapping left_kept_reads_seg2 to genome genome with Bowtie2 (2/12)
[2014-06-02 13:39:40] Mapping left_kept_reads_seg3 to genome genome with Bowtie2 (3/12)
[2014-06-02 13:40:11] Mapping left_kept_reads_seg4 to genome genome with Bowtie2 (4/12)
[2014-06-02 13:40:42] Mapping left_kept_reads_seg5 to genome genome with Bowtie2 (5/12)
[2014-06-02 13:41:13] Mapping left_kept_reads_seg6 to genome genome with Bowtie2 (6/12)
[2014-06-02 13:41:43] Mapping left_kept_reads_seg7 to genome genome with Bowtie2 (7/12)
[2014-06-02 13:42:15] Mapping left_kept_reads_seg8 to genome genome with Bowtie2 (8/12)
[2014-06-02 13:42:46] Mapping left_kept_reads_seg9 to genome genome with Bowtie2 (9/12)
[2014-06-02 13:43:16] Mapping left_kept_reads_seg10 to genome genome with Bowtie2 (10/12)
[2014-06-02 13:43:46] Mapping left_kept_reads_seg11 to genome genome with Bowtie2 (11/12)
[2014-06-02 13:44:16] Mapping left_kept_reads_seg12 to genome genome with Bowtie2 (12/12)
[2014-06-02 13:44:46] Searching for junctions via segment mapping
[2014-06-02 13:47:10] Retrieving sequences for splices
[2014-06-02 13:49:33] Indexing splices
[2014-06-02 13:49:34] Mapping left_kept_reads_seg1 to genome segment_juncs with Bowtie2 (1/12)
[2014-06-02 13:49:40] Mapping left_kept_reads_seg2 to genome segment_juncs with Bowtie2 (2/12)
[2014-06-02 13:49:44] Mapping left_kept_reads_seg3 to genome segment_juncs with Bowtie2 (3/12)
[2014-06-02 13:49:49] Mapping left_kept_reads_seg4 to genome segment_juncs with Bowtie2 (4/12)
[2014-06-02 13:49:53] Mapping left_kept_reads_seg5 to genome segment_juncs with Bowtie2 (5/12)
[2014-06-02 13:49:57] Mapping left_kept_reads_seg6 to genome segment_juncs with Bowtie2 (6/12)
[2014-06-02 13:50:02] Mapping left_kept_reads_seg7 to genome segment_juncs with Bowtie2 (7/12)
[2014-06-02 13:50:06] Mapping left_kept_reads_seg8 to genome segment_juncs with Bowtie2 (8/12)
[2014-06-02 13:50:10] Mapping left_kept_reads_seg9 to genome segment_juncs with Bowtie2 (9/12)
[2014-06-02 13:50:14] Mapping left_kept_reads_seg10 to genome segment_juncs with Bowtie2 (10/12)
[2014-06-02 13:50:18] Mapping left_kept_reads_seg11 to genome segment_juncs with Bowtie2 (11/12)
[2014-06-02 13:50:22] Mapping left_kept_reads_seg12 to genome segment_juncs with Bowtie2 (12/12)
[2014-06-02 13:50:26] Joining segment hits
[2014-06-02 13:52:51] Reporting output tracks
[FAILED]
Error running /usr/bin/tophat_reports --min-anchor 8 --splice-mismatches 0 --min-report-intron 50 --max-report-intron 500000 --min-isoform-fraction 0.15 --output-dir /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq// --max-multihits 20 --max-seg-multihits 40 --segment-length 25 --segment-mismatches 2 --min-closure-exon 100 --min-closure-intron 50 --max-closure-intron 5000 --min-coverage-intron 50 --max-coverage-intron 20000 --min-segment-intron 50 --max-segment-intron 500000 --read-mismatches 2 --read-gap-length 2 --read-edit-dist 2 --read-realign-edit-dist 3 --max-insertion-length 3 --max-deletion-length 3 -z gzip -p8 --no-closure-search --no-coverage-search --no-microexon-search --sam-header /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//tmp/genome_genome.bwt.samheader.sam --report-discordant-pair-alignments --report-mixed-alignments --samtools=/usr/bin/samtools --bowtie2-max-penalty 6 --bowtie2-min-penalty 2 --bowtie2-penalty-for-N 1 --bowtie2-read-gap-open 5 --bowtie2-read-gap-cont 3 --bowtie2-ref-gap-open 5 --bowtie2-ref-gap-cont 3 /home/koen/Host/Stage_Enschede/methods_Bowtie2/indexes_chromFA/genome.fa /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//junctions.bed /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//insertions.bed /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//deletions.bed /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//fusions.out /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//tmp/accepted_hits /home/koen/Host/Stage_Enschede/data_RNA-SampleA3/3_S3_L001_R1_001.fastq//tmp/left_kept_reads.bam
Loading ...done | Problem solved. It wasn't due to the RAM but due to the `-p 8`. Apparently it only works with `-p 1`. | biostars | {"uid": 102197, "view_count": 4037, "vote_count": 2} |
I am trying to test for over-representation of a set of overlapped chip-seq peaks between constitutive exons and alternatively spliced exons.
I have a bed file that contains my overlapped chip peaks, a bed file that contains constitutive exons and a bed file that contains alternatively spliced exons (generated using a custom script provided by some authors of a paper). I am interested in running a statistical test that tells me whether my overlapped peaks are over-represented / enriched in my constitutive exon file, or alternatively spliced exon file individually.
I thought about running a hypergeometric test using the phyper function in R. But I'm not quite sure what numbers I would use specifically.
I also attempted to use the bedtools fisher test by using my overlapped chip-seq peak file and testing that against my con exon file and then my alt exon file seperately. This returned a p-value of 0 for both which I guess doesn't make much sense (though I am not very math-oriented). I mostly work on wet-lab stuff as an assistant.
Any help is appreciated. | You can use the R/Bioconductor package [regioneR][1] for this. It implements a statistical test for the association of genomic regions (such as chip peaks and exons) based on random permutations.
In this case I think the best aproach would be to "flip" the question and ask whether alternatively spliced (or constitutive) exons tend to be associated with the chip peaks and use the "resampling" randomization strategy.
For example (untested code!)
library(regioneR)
chip.peaks <- toGRanges("chip.peaks.bed")
alt.exons <- toGRanges("alt.exons.bed")
const.exons <- toGRanges("const.exons.bed")
all.exons <- c(alt.exons, const.exons)
pt <- permTest(A=alt.exons, B=chip.peaks, universe=all.exons,
randomize.function = resampleRegions, evaluate.function = numOverlaps,
ntimes = 1000)
pt
plot(pt)
This will create 1000 random sets of exons and test if the alt.exons are more associated with the peaks than one could expect by chance.
You can find more information about how to use regioneR and about permutation tests in the [package vignette][2].
[1]: https://bioconductor.org/packages/release/bioc/html/regioneR.html
[2]: https://bioconductor.org/packages/release/bioc/vignettes/regioneR/inst/doc/regioneR.pdf | biostars | {"uid": 167359, "view_count": 2438, "vote_count": 1} |
<p>Dear all,</p>
<p>I have checked many post in biostar and other blogs to find orthologous and paralogous gene identification across/within the species. Most of them are suggesting for Best Reciprocal Blast Method(BBRM). But i have 2 leaf and 2 rhizome transcript samples. I have to find out orthologous and paralogous genes between these set. Shall i use BRBM method or are there any other better way to do it.</p>
<p>Please suggest any tools.</p>
<p>Thanks all</p>
| <p>Thanks for the suggestion.</p>
<p>If i am not mistaken, you are suggesting BRBM (Best Reciprocal Blast method) to use. If so i will find orthologous genes between samples. Could you please suggest any way to find paralogous genes as well</p>
| biostars | {"uid": 121426, "view_count": 3790, "vote_count": 3} |
Hello,
I have a couple of bam files from **exome sequencing** and a list of regions that I want to perform variant calling and variant annotation on. My goal is to look for evidence of mutations in those regions that go beyond canonical LOF protein coding mutations.
I am thinking of extracting the regions of interest from a bam, converting to a vcf and then annotating it using snpEff or something like that. The following is my commandline:
# add read groups
picard AddOrReplaceReadGroups I=sample.bam O=sample.fixed.bam RGID=4 RGLB=lib1 RGPL=illumina RGPU=unit1 RGSM=20
# run gatk
java GenomeAnalysisTK.jar -T HaplotypeCaller -R human_g1k_v37.fasta
-I sample.fixed.bam --emitRefConfidence GVCF --variant_index_type LINEAR --variant_index_parameter 128000 --genotyping_mode DISCOVERY -stand_emit_conf 10 -stand_call_conf 30 -o sample.gvcf
# run snpeff
snpeff -c snpEff.GRCh37.config -ud 10 -classic GRCh37.75 sample.gvcf > sample.snpeff.gvcf
Q1. Is my approach correct? Is this approach applicable to WGS and RNASeq as well?
My vcf file generated in this manner has missing ALT info - it says <NON_REF>. How can I fix this?
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 20
chr4 54243811 . C <NON_REF> . . END=54243904 GT:DP:GQ:MIN_DP:PL 0/0:208:99:129:0,120,1800
chr4 54243905 . T C,<NON_REF> 10162.77 . DP=280;MLEAC=2,0;MLEAF=1.00,0.00;MQ=59.94 GT:AD:DP:GQ:P
L:SB 1/1:0,268,0:268:99:10191,805,0,10191,805,10191:0,0,160,108
chr4 54243906 . C <NON_REF> . . END=54244236 GT:DP:GQ:MIN_DP:PL 0/0:170:99:37:0,99,1485
chr4 54244237 . T <NON_REF> . . END=54244238 GT:DP:GQ:MIN_DP:PL 0/0:36:90:35:0,90,1350
chr4 54244239 . C <NON_REF> . . END=54244241 GT:DP:GQ:MIN_DP:PL 0/0:34:72:34:0,72,1080
chr4 54244242 . G <NON_REF> . . END=54244244 GT:DP:GQ:MIN_DP:PL 0/0:33:63:33:0,63,945
chr4 54244245 . C <NON_REF> . . END=54244247 GT:DP:GQ:MIN_DP:PL 0/0:41:78:39:0,78,1170
Q2. Also, can someone suggest a better approach for creating a vcf for just specific regions of bam file? | You can use the -L flag of HaplotypeCaller to limit the intervals in which variants are detected as specified by a bed file.
> --intervals / -L One or more genomic intervals over which to operate Use this option to perform the analysis over only part of the genome.
> This argument can be specified multiple times. You can use
> samtools-style intervals either explicitly on the command line (e.g.
> -L chr1 or -L chr1:100-200) or by loading in a file containing a list of intervals (e.g. -L myFile.intervals). Additionally, you can also
> specify a ROD file (such as a VCF file) in order to perform the
> analysis at specific positions based on the records present in the
> file (e.g. -L file.vcf). Finally, you can also use this to perform the
> analysis on the reads that are completely unmapped in the BAM file
> (i.e. those without a reference contig) by specifying -L unmapped.
from https://software.broadinstitute.org/gatk/gatkdocs/org_broadinstitute_gatk_engine_CommandLineGATK.php | biostars | {"uid": 208412, "view_count": 2138, "vote_count": 1} |
I'm working with drugbank ids and I'd like to incorporate adverse effects from OFFSIDES database http://tatonettilab.org/offsides/. I'll appreciate help in mapping between the ids. | looks like NIH applies RxNorm ids to drugbank, maybe use their mapping?
https://www.nlm.nih.gov/research/umls/rxnorm/sourcereleasedocs/drugbank.html | biostars | {"uid": 9494449, "view_count": 796, "vote_count": 1} |
Hello All,
I am trying to reanalyze a WGS dataset that was generated a few years ago. I've access to the old BAM files and I was able to create paired fastq's for each sample. Since I'll be using picard for marking duplicates I would like to add the read group information at the time of aligning my fastq's with BWA mem. From the old bam file, I was able to get lines matching the '@RG' lines. But there seems to be multiple read group ID present in the bam file. From the BWA documentation, it seems that the correct way of adding the read group info is by **bwa mem -R '@RG\tID:foo\tSM:bar\tLB:library1'**. I believe here both foo and bar are unique for each sample. For my particular case, how should multiple read group info to BWA? Hope the question is clear. I've very limited experience with WGS. I appreciate all your help and comments.
samtools view -H sampleA.bam | grep '^@RG'
@RG ID:AVKMG.3 SM:sampleA LB:0993462810_Illumina PL:ILLUMINA PU:AVKMGDSXX191015.3.GTCCACAG-CGCGAATA CN:BI DT:2016-10-15T04:00:00+0000 DS:KS-9108
@RG ID:AJJMK.4 SM:sampleA LB:0993462810_Illumina PL:ILLUMINA PU:AJJMKDSXX191014.4.GTCCACAG-CGCGAATA CN:BI DT:2016-10-14T04:00:00+0000 DS:KS-9108
@RG ID:AKKMD.4 SM:sampleA LB:0993462810_Illumina PL:ILLUMINA PU:AKKMDDSXX191014.4.GTCCACAG-CGCGAATA CN:BI DT:2016-10-14T04:00:00+0000 DS:KS-9108
@RG ID:UGGMD.4 SM:sampleA LB:0993462810_Illumina PL:ILLUMINA PU:UGGMDDSXX191014.4.GTCCACAG-CGCGAATA CN:BI DT:2016-10-14T04:00:00+0000 DS:KS-9108
| Most likely the alignments have been done separately and bam files merged afterwards. | biostars | {"uid": 481666, "view_count": 962, "vote_count": 2} |
Hi,
I have PDX RNA-seq data.I want to separate the reads of mouse and human,before comparing with tumor RNA-seq data. Is there any tool to do that?
After removing the mouse reads I want to do differential expression between primary tumors and PDX tumors.
Thanks,
Ron | [BBSplit][1] will map the reads simultaneously to multiple references, and output in multiple files, one per reference. Since you're looking at RNA-seq of tumors, you would want to increase the sensitivity over the default, using settings like (if mapping to the genomes):
bbsplit.sh in=reads.fq ref=hg19.fa,mm10.fa minratio=0.5 maxindel=100000 minhits=1 basename=out_%.fq ambig2=all local
If mapping to transcriptomes, `maxindel=500` would probably be better than `100000`.
[1]: https://sourceforge.net/projects/bbmap/ | biostars | {"uid": 143019, "view_count": 10234, "vote_count": 4} |
Hello. As I am trying to download genomes from NCBI using command [centrifuge-download], a warning keeps popping up: "line 24: dustmasker: command not found." The downloaded files end up with 0 Kb and all the subsequent steps are unperformable. The version is centrifuge-1.0.3-beta and the command is as follow:
centrifuge-download -o library -m -d "archaea,bacteria,viral" refseq > seqid2taxid.map
Anyone helps me and saves my life :(
| Or download a dustmasker binary for your system [here][1] - it is part of the BLAST+ package
[1]: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ | biostars | {"uid": 251868, "view_count": 3770, "vote_count": 1} |
<p>Hi all,</p>
<p>Can you tell me what's the meaning of <strong>@RG</strong> / <strong>group</strong> in the <strong>SAM</strong> format. Can I consider it as the very same thing as '<strong>Sample-name</strong>' ?</p>
<p>Is there a case where one could find more than one <strong>@RG</strong> tag in a <strong>BAM</strong> file ?</p>
<p>Can <strong>mpileup</strong> (or another software) use this <strong>@RG</strong> flag to put more than one sample column in a <strong>VCF</strong> file.</p>
<p>If yes, what does it mean for the <strong>QUAL</strong> column ?</p>
<p>If no, under which condition can we find more than one sample column in a VCF ?</p>
<p>Thanks,</p>
<p>Pierre</p>
| <p>For Illumina reads, RG typically groups reads from a lane. GATK requires/assumes such a use in several components. At least one should not put reads from multiple libraries in the same RG.</p>
<p>When reading the input BAMs, <a href='http://samtools.sourceforge.net/'>samtools</a> tries to group reads based on their @RG-SM tag. If there are no @RG lines, it implicitly inserts the following line into the header:</p>
<pre><code>@RG ID:filename SM:filename
</code></pre>
<p>The VCF spec is very clear about the definition of QUAL: the probability of there being no SNPs in any samples.</p>
| biostars | {"uid": 9724, "view_count": 14937, "vote_count": 7} |
Hi,
I am trying to install FASTX toolkit by following the instructions from their website. http://hannonlab.cshl.edu/fastx_toolkit/install_ubuntu.txt
While installing libgtextutils, make command gives following error:
make all-recursive
make[1]: Entering directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6'
Making all in m4
make[2]: Entering directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/m4'
make[2]: Nothing to be done for 'all'.
make[2]: Leaving directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/m4'
Making all in src
make[2]: Entering directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/src'
Making all in gtextutils
make[3]: Entering directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/src/gtextutils'
/bin/bash ../../libtool --tag=CXX --mode=compile g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT stream_wrapper.lo -MD -MP -MF .deps/stream_wrapper.Tpo -c -o stream_wrapper.lo stream_wrapper.cpp
libtool: compile: g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT stream_wrapper.lo -MD -MP -MF .deps/stream_wrapper.Tpo -c stream_wrapper.cpp -fPIC -DPIC -o .libs/stream_wrapper.o
libtool: compile: g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT stream_wrapper.lo -MD -MP -MF .deps/stream_wrapper.Tpo -c stream_wrapper.cpp -o stream_wrapper.o >/dev/null 2>&1
mv -f .deps/stream_wrapper.Tpo .deps/stream_wrapper.Plo
/bin/bash ../../libtool --tag=CXX --mode=compile g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT text_line_reader.lo -MD -MP -MF .deps/text_line_reader.Tpo -c -o text_line_reader.lo text_line_reader.cpp
libtool: compile: g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT text_line_reader.lo -MD -MP -MF .deps/text_line_reader.Tpo -c text_line_reader.cpp -fPIC -DPIC -o .libs/text_line_reader.o
libtool: compile: g++ -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -DDEBUG -g -O1 -MT text_line_reader.lo -MD -MP -MF .deps/text_line_reader.Tpo -c text_line_reader.cpp -o text_line_reader.o >/dev/null 2>&1
mv -f .deps/text_line_reader.Tpo .deps/text_line_reader.Plo
/bin/bash ../../libtool --tag=CC --mode=compile gcc -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -MT strnatcmp.lo -MD -MP -MF .deps/strnatcmp.Tpo -c -o strnatcmp.lo strnatcmp.c
libtool: compile: gcc -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -MT strnatcmp.lo -MD -MP -MF .deps/strnatcmp.Tpo -c strnatcmp.c -fPIC -DPIC -o .libs/strnatcmp.o
libtool: compile: gcc -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -MT strnatcmp.lo -MD -MP -MF .deps/strnatcmp.Tpo -c strnatcmp.c -o strnatcmp.o >/dev/null 2>&1
mv -f .deps/strnatcmp.Tpo .deps/strnatcmp.Plo
/bin/bash ../../libtool --tag=CC --mode=compile gcc -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -MT pipe_fitter.lo -MD -MP -MF .deps/pipe_fitter.Tpo -c -o pipe_fitter.lo pipe_fitter.c
libtool: compile: gcc -DHAVE_CONFIG_H -I. -I../.. -g -O2 -Wall -Wextra -Wformat-nonliteral -Wformat-security -Wswitch-default -Wswitch-enum -Wunused-parameter -Wfloat-equal -Werror -DDEBUG -g -O1 -MT pipe_fitter.lo -MD -MP -MF .deps/pipe_fitter.Tpo -c pipe_fitter.c -fPIC -DPIC -o .libs/pipe_fitter.o
pipe_fitter.c: In function ‘pipe_close’:
pipe_fitter.c:30:6: error: variable ‘i’ set but not used [-Werror=unused-but-set-variable]
int i, status ;
^
cc1: all warnings being treated as errors
Makefile:336: recipe for target 'pipe_fitter.lo' failed
make[3]: *** [pipe_fitter.lo] Error 1
make[3]: Leaving directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/src/gtextutils'
Makefile:235: recipe for target 'all-recursive' failed
make[2]: *** [all-recursive] Error 1
make[2]: Leaving directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6/src'
Makefile:311: recipe for target 'all-recursive' failed
make[1]: *** [all-recursive] Error 1
make[1]: Leaving directory '/media/wkstn/Data/Course/Project/libgtextutils-0.6'
Makefile:220: recipe for target 'all' failed
make: *** [all] Error 2
Is anyone familiar with how I can fix this?
| The problem comes from here:
pipe_fitter.c: In function ‘pipe_close’:
pipe_fitter.c:30:6: error: variable ‘i’ set but not used [-Werror=unused-but-set-variable]
int i, status ;
but I'm suprised because as far as I can see https://github.com/agordon/libgtextutils/blob/master/src/gtextutils/pipe_fitter.c#L48 'i' is declared and used here: https://github.com/agordon/libgtextutils/blob/master/src/gtextutils/pipe_fitter.c#L35
is it the same code you see in your source ?
src/gtextutils/pipe_fitter.c
are you using the latest version ? you should download the source from github. ?
| biostars | {"uid": 244378, "view_count": 4823, "vote_count": 2} |
Hi,
Is any tool/method to convert article title to pubmed ID? Anyone know?
Thank you,
| from lxml import etree
from requests import get
import sys
import os
url_for_get_id = "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=pubmed&field=title&term=\"{}\""
url_for_get_title = "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=pubmed&id={}&retmode=XML"
def xml_id_or_title_extractor(page):
"""extract the ids from page and return that as string"""
content = etree.fromstring(page)
root = content.getroottree()
result = [item.text for item in root.xpath(xpath)]
if len(result)<1:
return "Not Found!!"
return ",".join(result)
def write_to_file(_list):
result = open("title_pubmedid_result.txt","a")
for title in _list:
try:
title = title.strip()
page = get(URL.format(title)).content
_id = xml_id_or_title_extractor(page)
result.write(_id + "\t" + title + "\n")
print (title,"===>","OK")
except:
print("An Exception occurred!! on \n <{}>".format(title))
result.write("ERROR" + "\t" + title + "\n")
result.close()
def run(title_list):
if len(sys.argv) > 2:
path = os.getcwd()
file_path = path+"/"+sys.argv[2]
try:
read_file = [item.strip() for item in open(file_path).readlines()]
write_to_file(read_file)
except FileNotFoundError:
print ("your file is not exsists or input file not in same directory")
except:
print ("there is a problem with your input file")
else:
write_to_file(title_list)
if __name__ == "__main__":
print ("the script is running\n")
print ("this is a script to convert article paper to pubmedid and vice versa")
print ("*"*20)
if len(sys.argv)<2:
print ("\nyour arguments are not enough !!\n \n Use this format : for example \n\n< python pubmedid_to_title.py -t2id your_file.txt> for reading from file\n\n\
or \n\n< python pubmedid_to_title.py -t2id> for reading from a list")
sys.exit()
elif sys.argv[1] == "-t2id":
URL = url_for_get_id
xpath = "//Id"
_id_or_title_list = ["Differentially Expressed miRNAs in Hepatocellular Carcinoma Target Genes in the Genetic Information Processing and Metabolism Pathways.",
"miR-429 inhibits migration and invasion of breast cancer cells in vitro."]
elif sys.argv[1] == "-id2t":
URL = url_for_get_title
xpath = "//ArticleTitle"
_id_or_title_list = ["18456660","19196975"]
else:
print ("use -id2t for convert id to title or -t2id for convert title to id as first argument!! ")
sys.exit()
# suppose you have a list of paper's title, for example two titles or two id ==> _id_or_title_list list
# you can edit it ...
# if you wnat to read from a file, your file name come after it, be careful the script and file directory must be in same directory
# also each line should be just one title
# for example type this:
# python title_to_pubmedid.py -t2id your_file.txt
# first argument must be -t2id for convert titles to ides and -id2t for reverse search
#SO YOU HAVE TWO CHICE: 1:READ FROM FILE 2:EDIT THE ABOVE LIST :D
run(_id_or_title_list)
| biostars | {"uid": 190968, "view_count": 4679, "vote_count": 1} |
Can I download gene expression data from ICGC? If so, how? Do I need controlled access? | Gene expression data is open. Not sure if that's what you need but you can download it the following way:
On Cancer Projects page on the left panel check EXP-S (Sequencing-based Gene Expression) and/or EXP-A (Array-based Gene Expression) in "Available Data Type section". You'll see all projects for which specified data type(s) are available ([example][1])
Then you can download expression data for all donors of one project through data repository: for example current data release has .tsv exp_array.BRCA-US.tsv.gz and exp_seq.BRCA-US.tsv.gz for [BRCA-US project][2]
You can also download gene expression data for one donor: on Donors page apply the same filtering as previously metioned ([example][3]). When you click on donor ID you'll be taken to donors page where you can click "Download Donor Data" button and select the type(s) of data you want.
Hope this helps.
[1]: https://dcc.icgc.org/projects/details?filters=%7B%22project%22:%7B%22availableDataTypes%22:%7B%22is%22:[%22exp_seq%22,%22exp_array%22]%7D%7D%7D
[2]: https://dcc.icgc.org/releases/current/Projects/BRCA-US
[3]: https://dcc.icgc.org/search?filters=%7B%22donor%22:%7B%22availableDataTypes%22:%7B%22is%22:[%22exp_seq%22,%22exp_array%22]%7D%7D%7D | biostars | {"uid": 184539, "view_count": 4927, "vote_count": 2} |
I have access to some indexed BAM files in S3. Using the [AWS CLI][1], it is fairly easy to download the entire BAM file. That was fine for our initial analysis, but for validation, we are interested in looking a small region of ~10kb in thousands of individuals.
The BAM files are indexed and S3 supports GET requests with [range headers][2] so this should be possible. Does anyone know of a tool that does this?
EDIT: [htslib 1.3][3] was recently released and supports random access to BAM files in s3.
[1]: http://docs.aws.amazon.com/cli/latest/reference/s3/cp.html
[2]: https://docs.aws.amazon.com/AmazonS3/latest/dev/GettingObjectsUsingAPIs.html
[3]: https://github.com/samtools/htslib/releases/tag/1.3 | The upcoming samtools 1.3 release will support this. As well as the current way of accessing public buckets in donfreed's comment on another answer, samtools 1.3 will understand s3: pseudo-URLs like
s3://1000genomes/phase1/data/NA12878/exome_alignment/NA12878.blah.bam
For accessing private buckets, samtools will look for your AWS credentials in the usual configuration files and environment variables, or you can specify them on the command line as *s3://id:secret@bucket/*... though that's not particularly recommended.
This release will be fairly soon. In the meantime, you can try this out by building samtools with GitHub htslib's **libcurl** branch. The code in that branch only looks for credentials in `$AWS_ACCESS_KEY_ID` / `$AWS_SECRET_ACCESS_KEY` and as *id:secret* in the URL. | biostars | {"uid": 147772, "view_count": 9353, "vote_count": 4} |
I found some tips for speeding up `read.table()` [here][1], I wondered if anyone can suggest something for [read.fasta()][2], which is part of the seqinr library. Not that they're really comparable.
I have a ~6GB file.
[1]: http://www.biostat.jhsph.edu/~rpeng/docs/R-large-tables.html
[2]: http://www.inside-r.org/packages/cran/seqinr/docs/read.fasta | If you have a huge file (just reread your post, 6Gb), the best way is to use an index (this is how databases work...). Take a look at this link: http://stackoverflow.com/questions/23173215/how-to-subset-sequences-in-fasta-file-based-on-sequence-id-or-name
It depends on what you want to do; do you only need a subset of these sequences at any given time or do you require every one of them? You would need a way to load and store them in your RAM, rather than reading a 6Gb file each time you tinker your file. NB: you can increase R's memory to avoid errors. | biostars | {"uid": 143485, "view_count": 8389, "vote_count": 1} |
Hi,
When I am asked to mention for which I have recently used a programming language like Python in data analysis, I just can say that I used Python once to extract some genome coordinates from gene bank. But definitely this is not a wise answer and disappointing for a bioinformatician. How then a medium data analyst could use Python? I mean for which purpose as a good answer.
Thank you very much
| If this is hypothetical (and not explicitly about how *you* have used Python) then you can say that Python can be used for pretty much everything, although it has it's strong points and weaker points.
You could use python to create a pipeline (snakemake), make plots (matplotlib, seaborn), interface with databases (sqlite), perform processing of fasta/fastq (biopython) and create websites (django).
But even more important than knowing what you can is knowing the limitations. While you have a lot of statistical modules in Python - usually R is the more appropriate language here, definitely for differential expression analysis. But there is nothing that you can do in R that you cannot do in Python. For making production ready code for which speed is an issue then Python is often not your best guess, rather something compiled such as C or Java, or more recent languages such as Go or Rust. It's often 1) having a language you are comfortable in and 2) using the right tool for the job at hand.
I really dislike R, but I do differential expression analysis in R. | biostars | {"uid": 282001, "view_count": 1076, "vote_count": 1} |
Hello,
I'm analyzing TCGA breast cancer data to classify the samples into their respective subtypes, ad then to check if the genes of our study have a subtype specific pattern of expression. To do this, I was suggested to use `genefu`. At the step of classifying the subtypes -
`PAM50Base <- molecular.subtyping(sbt.model = "pam50",data=data, annot=annot,do.mapping=F)`
I get an error -
`Error in intrinsic.cluster.predict(sbt.model = pam50.robust, data = data, : `
`no probe in common -> annot or mapping parameters are necessary for the mapping process!`
In the command, annot is the file used for annotation and is of the format -
>probe EntrezGene.ID Gene.ID Gene.Symbol
Data refers to the input file, which is of the format -
>Gene.Symbol Sample1 SAMPLE2 ... Sample 1092
Both files are tab delimited. I want to know if anyone has done this before, and if the file formatting is correct?
P.S. I have tried using the `Gene.Symbol` and `probe` in the data file, but both give the same error.
Edit: Should my data file also contain the `EntrezGene.ID` column?
Thank you. | Hi vinayjrao,
I think the problem is with the `data` matrix; `molecular.subtyping` fuction expects a matrix of samples(rows) x genes(cols). As I can see above your data matrix is genes x samples, right? Try transposing the matrix.
From the genefu vignette for do.mapping: `TRUE if the mapping through Entrez Gene ids must be performed (in case of ambiguities, the most variant probe is kept for each gene)` | biostars | {"uid": 313540, "view_count": 4488, "vote_count": 2} |
Hello,
In our group we are about to design an amplicon-based targeted panel for NGS, and we're aiming at around 150 bp for mean amplicon sizes. The wet lab people came back to me asking to calculate the optimal number of samples to be loaded in the instrument (MiSeq). And this is where I have my question: ideally I'd like to pick 2x150bp as read length, so I can have a greater output, however the mean amplicon size might be close to the read length, or in some cases, lower.
Hence my question: would it be "safe" to use a read length approximately equal to the amplicon length so that it doesn't complicate my analysis job afterwards (I'm thinking about adapter read through)? My previous experience always had a larger amplicon length (> 200 bp) so it can't really compare.
Should I still go for 2x150bp, or aim lower at 2x75bp? If I use 2x150bp, would possible adapter read through throw a spanner in the works, or should soft clipping (from BWA) be enough? | I'd still do 2x150 if the amplicon targets are 150bp on average. You can deal with the adapter read through and trimming fairly easily. Something like 200bp averaged amplicons might be a better size overall, but either way I think is fine. | biostars | {"uid": 183992, "view_count": 4880, "vote_count": 1} |
Trying to understand substitution matrices. It seems like it is a scoring scheme for alignments, particularly if you are looking for homology?
I am trying to see if it would be applicable if I am looking at mutations between proteins from different people. Since my sequences are very similar with only 1 or 2 mutations between them, the substitution matrix would probably not be applicable here? I am assuming if there is a nonsynonymous mutation between two sequences it would give me a score (say BLOSUM62) based on how likely that substitution would occur in nature? Are there other ways to interpret these scoring matrices? | Before we go into your question, it may be best and most concise to simply describe the exact SNP sites and leave it at that, given that your proteins are so similar. However, here are the differences in PAM and BLOSUM:
BLOSUM (BLOcks SUbstitution Matrix) were derived by looking at alignments of highly conserved protein domains at different evoluntionarily divergent distances, then taking into account how frequently one amino acid was substituted to another. It's described in [this paper by Henikoff][1]. They are based on local alignment of conserved protein regions.
PAM (Point Accepted Mutations) matrices were first described by Margaret Dayhoff (who was a fantastic scientist, even in face of the challenges of her role given the time period). "Each entry in a PAM matrix indicates the likelihood of the amino acid of that row being replaced with the amino acid of that column through a series of one or more point accepted mutations during a specified evolutionary interval, rather than these two amino acids being aligned due to chance." They are based on global alignment.
In short, this is what matters about the differences between the two:
1. PAM matrices are typically used on more closely related proteins (such as your case), BLOSUM are typically used on more evolutionarily divergent proteins.
2. The greater the PAM number the more DISTANT the sequences being compared should be; the greater the BLOSUM number, the more SIMILAR the sequences being compared should be.
So for your application, if you were to use these, you should either use a LOW PAM matrix or a HIGH BLOSUM matrix number. Whether this is appropriate for your application depends on what you want to get out of it (e.g. the whole protein difference or just local protein domain differences); you're right in that they are typically used for alignment scoring, but they can also be used to generate some evolutionary cost distance. However, there may be better methods out there for your purpose if you look for methods for creating distance trees based on some metric.
[1]: https://www.ncbi.nlm.nih.gov/pubmed/1438297 | biostars | {"uid": 190423, "view_count": 2059, "vote_count": 1} |
Hi there,
I always used TCGAbiolinks to get raw count for TCGA projects like below:
expquery <- GDCquery(project = "TCGA-KIRC",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts")
GDCdownload(expquery,directory = "GDCdata")
expquery2 <- GDCprepare(expquery,directory = "GDCdata",summarizedExperiment = T)
expMatrix <- TCGAanalyze_Preprocessing(expquery2)
However, it does not work today and it seems there is no HTSeq - Counts
Error in GDCquery(project = "TCGA-KIRC", data.category = "Transcriptome Profiling", :
Please set a valid workflow.type argument from the list below:
=> STAR - Counts
Therefore, I used STAR - Counts to download the data, which has completely different format of what I downloaded before using HTSeq - Count. The expression matrix for each sample has more columns including fpkm_unstranded and tpm_unstranded.
Actually the data downloaded using STAR - Counts is much more useful but I do not know how to extract the files to a readable expression matrix (ENSEMBL ID as rownames and TCGA tumor barcode as column names) because the GDCprepare() function fails to work on it:
expquery2 <- GDCprepare(expquery,directory = "GDCdata",summarizedExperiment = T)
| | 0%
Error in readr::read_tsv(file = f, col_names = TRUE, progress = FALSE, :
unused argument (show_col_types = FALSE)
Error in if (value == n) { : argument is of length zero
Anyone has any solutions? Many thanks in advance! | ```
projects <- c("TCGA-LGG","TCGA-GBM")
ids <- c("TCGA-76-6661", "TCGA-26-5132", "TCGA-19-1389")
# RNA Transcriptome
query <- GDCquery(
project = projects,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts",
barcode = ids
)
GDCdownload(query,
method = "api",
directory = "./data",
files.per.chunk = 10)
rna <- GDCprepare(query, save = FALSE, summarizedExperiment = T, directory = "./data", remove.files.prepared = F)
rna <- as.data.frame(rna@assays@data@listData)
# DNA Meth Beta Value
query <-GDCquery(project = projects,
data.category= "DNA Methylation",
platform = "Illumina Human Methylation 450",
data.type = "Methylation Beta Value",
legacy = FALSE,
barcode = ids)
GDCdownload(query,
method = "api",
directory = "./data",
files.per.chunk = 10)
met <- GDCprepare(query, save = FALSE, summarizedExperiment = T, directory = "./data", remove.files.prepared = FALSE)
# IDAT Files
query <-GDCquery(project = projects,
data.category = "Raw microarray data",
data.type = "Raw intensities",
experimental.strategy = "Methylation array",
legacy = TRUE,
file.type = ".idat",
platform = "Illumina human methylation 450",
barcode = ids)
GDCdownload(query,
method = "api",
directory = "./data",
files.per.chunk = 10)
``` | biostars | {"uid": 9516907, "view_count": 3765, "vote_count": 1} |
I love .gfa, but sometimes I have trouble to understand them.
I have used Flye with pacBio reads with defaults options to make a first shot at assemble a linear bacterial genome. The genome probably contains some plasmids or some phages sequences. Flye gave me the following .gfa :
![enter image description here][1]
[1]: /media/images/4de3edca-7c14-4314-9dcc-e6f90b16
The green, yellow and red edges have been merged into one scaffold of around 9 Mb (as expected). And the little blue edge is his own contig of 42 Kb.
In his assembly_infot.txt file, Flye report that the big scaffold is indeed non linear, and that the tiny one is circular :
#seq_name length cov. circ. repeat mult. alt_group graph_path
scaffold_2 8926751 84 N N 1 * *,1,2,4,??,4,-3,-1,*
contig_4 42310 940 N Y 12 * 4
I have a few questions that puzzles me about this graph :
- Why does the .gfa connect all the edges into a circle if flye report that only one piece is circular not the other ?
- The yellow edge is connected by the same end to both green and red... And if the mean coverage of the green and red is 70X, it is only 17X for the yellow edge. What could it mean ? I am very puzzled by the fact it is connected by the same end. Could it be Flye trying to circularize it ? Or a sort of SV ? I think the DNA provided comes from a single colony so I don't see how that could be a SV.
- The blue edge has connection to itself, I imagine it is because of repetitions. But in the graph, this repetition is somehow connected to the others edges. So why was it split in the .fasta file in the end ?
| Oh, I forgot to update this post, but fenderglass, the author of flye, helped me a lot to understand this. You can follow the whole conversation here : https://github.com/fenderglass/Flye/issues/389 | biostars | {"uid": 9472019, "view_count": 930, "vote_count": 1} |
Hello Biostars,
What are an approximate quantity of human genes, transcripts (coding transcripts), proteins and metabolites? What about a model plant like Arabidopsis?
Thanks | Here are some statistics that I found in different databases:
For human:
- Coding genes: about 20,441
- Non-coding genes: about 22,219
- Transcription factors: about 2,067 (TFs/total genes= 10 %)
- Transcripts: about 198,002 (Transcripts/genes = 9.68)
- Proteins: more than 30,057
- Metabolites: more than 42162
Any revision or adding more information to this post would be
appreciated,
| biostars | {"uid": 241563, "view_count": 4984, "vote_count": 1} |
I want to get the information of all genes on human Y chromosome, then I found the statistics in different databases --Ensembl (GENCODE), NCBI, HGNC -- are dissimilar.
For example, protein-coding genes numbers:
CCDS 63
HGNC 45
Ensembl 63
NCBI 73
So what leads to these number be different?
By the way, is RefSeq gene data the same as NCBI homo sapiens annotation release? | The different resources you cite do different things and are not necessarily in sync. The CCDS tries to identify annotations of protein-coding regions in the human and mouse genomes that are consensual across several groups/institutes. The HGNC is in charge of attributing official names and symbols to genes. NCBI's RefSeq is a collection of sequences that are annotated as belonging to a gene and/or linked to other NCBI resources.
Ensembl provides a full genome annotation integrating many information types. Of the resources you cite, Ensembl is the only one that annotates the underlying genome.
When doing a bioinformatics project, select one reference and stick to it. Don't mix and match, this would be asking for trouble. I would recommend using Ensembl because it's much better organized and integrated than NCBI resources. | biostars | {"uid": 273735, "view_count": 1787, "vote_count": 1} |
I'm analyzing gene expression data from an experiment where I have (i) 6 different mice (A:D); (ii) 2 different time points ("pre" and "post" treatment); and (iii) 2 different response types ("responder" vs. "non-responder"). I'm trying to find genes with differential expression after treatment in the responders vs. non-responders. In other words: if I look at post therapy gene expression changes compared to 'pre', which genes are differentially expressed in responders compared to non-responders? Additionally, how can I include the 'paired' aspect into my analysis, where I might get more meaningful information by running the above analysis in a paired, rather than general, manner?
I've spent an entire day reading tutorials on designing design matrices for differential expression but I can't pin down the details, I'm hoping someone with experience might be able to help out.
A small reproducible example:
ms = factor(rep(c("A", "B", "C", "D", "E", "F"), each = 2)) # Mouse ID
rx = factor(rep(c("Pre", "Post"), 6)) # Pre or Post treatment
rp = factor(c(rep("NR", 6), rep("R", 6))) # Is the mouse a 'responder' or 'non-responder'?
data.frame(ms, rx, rp)
ms rx rp
1 A Pre NR
2 A Post NR
3 B Pre NR
4 B Post NR
5 C Pre NR
6 C Post NR
7 D Pre R
8 D Post R
9 E Pre R
10 E Post R
11 F Pre R
12 F Post R
The ideal analysis that I'd like to do is something like the following, but including the paired design, which would hopefully make the analysis more powerful:
(Post / Pre in responders) / (Post / Pre in non-responders)
The first term ("Post / Pre in responders") would find DE genes in responders, and by dividing by the second term ("Post / Pre in non-responders") I would ideally get genes which are diferentially expressed only in responders.
I tried running the "Post / Pre" analysis in responders and in non-responders separately, and manually dividing, as in the code above, but it doesn't feel right, and there must be a true way of running that with a properly designed matrix. | You'll want a design of `~ms + rx*rp` and the coefficient of primary interest is then the `rx:rp` interaction. That then accounts for the pairing and gives you the interaction you want. Note that you'll probably get an error about the matrix being of insufficient rank, in which case you should use `ms = factor(c(rep(rep(c("A","B","C"), each=2), 2))`. This will keep the pairing, but change things such that each of the groups contains a mouse with the same ID (otherwise correcting for the pairing will confound fitting the response group). | biostars | {"uid": 320399, "view_count": 1576, "vote_count": 1} |
Hi!
I'm trying to run fastQC on an HPC system but i'm having this error:
It seems that something is wrong with java.
Started analysis of raw_f.fastq
Approx 5% complete for raw_f.fastq
Approx 10% complete for raw_f.fastq
Approx 15% complete for raw_f.fastq
Approx 20% complete for raw_f.fastq
Approx 25% complete for raw_f.fastq
Approx 30% complete for raw_f.fastq
Approx 35% complete for raw_f.fastq
Approx 40% complete for raw_f.fastq
Approx 45% complete for raw_f.fastq
Approx 50% complete for raw_f.fastq
Approx 55% complete for raw_f.fastq
Approx 60% complete for raw_f.fastq
Approx 65% complete for raw_f.fastq
Approx 70% complete for raw_f.fastq
ecc...
javax.imageio.IIOException: Can't create cache file!
at javax.imageio.ImageIO.createImageInputStream(ImageIO.java:361)
at javax.imageio.ImageIO.read(ImageIO.java:1397)
at uk.ac.babraham.FastQC.Report.HTMLReportArchive.base64ForIcon(HTMLReportArchive.java:379)
at uk.ac.babraham.FastQC.Report.HTMLReportArchive.startDocument(HTMLReportArchive.java:303)
at uk.ac.babraham.FastQC.Report.HTMLReportArchive.<init>(HTMLReportArchive.java:84)
at uk.ac.babraham.FastQC.Analysis.OfflineRunner.analysisComplete(OfflineRunner.java:185)
at uk.ac.babraham.FastQC.Analysis.AnalysisRunner.run(AnalysisRunner.java:123)
at java.lang.Thread.run(Thread.java:745)
Caused by: java.nio.file.AccessDeniedException: /tmp/imageio6327552574161602354.tmp
at sun.nio.fs.UnixException.translateToIOException(UnixException.java:84)
at sun.nio.fs.UnixException.rethrowAsIOException(UnixException.java:102)
at sun.nio.fs.UnixException.rethrowAsIOException(UnixException.java:107)
at sun.nio.fs.UnixFileSystemProvider.newByteChannel(UnixFileSystemProvider.java:214)
at java.nio.file.Files.newByteChannel(Files.java:361)
at java.nio.file.Files.createFile(Files.java:632)
at java.nio.file.TempFileHelper.create(TempFileHelper.java:138)
at java.nio.file.TempFileHelper.createTempFile(TempFileHelper.java:161)
at java.nio.file.Files.createTempFile(Files.java:897)
at javax.imageio.stream.FileCacheImageInputStream.<init>(FileCacheImageInputStream.java:102)
at com.sun.imageio.spi.InputStreamImageInputStreamSpi.createInputStreamInstance(InputStreamImageInputStreamSpi.java:69)
I installed the latest fastQC version inside the HPC on my personal conda environment but I'm still getting the same error.
Fastqc creates the report.txt file but the HTML file is missed and i can't see any plot in the zipped file generated.
Any suggestion?
| your problem is here:
> Caused by: java.nio.file.AccessDeniedException: /tmp/imageio6327552574161602354.tmp
use option -d
-d --dir Selects a directory to be used for temporary files written when
generating report images. Defaults to system temp directory if
not specified. | biostars | {"uid": 482381, "view_count": 1017, "vote_count": 1} |
When analyzing clinical genomes, I often find it hard to visualize the genomic context of a potential driver mutation (say a point mutation on a key cancer gene). For example, is there simultaneous LOH/amplification at the mutation locus? How many copies of the mutant allele are there? Did the mutation arrive before/after the overlapping copy number change? Is there a structural variation breakpoint near by? Are there other point mutations close by?
Thus, I wonder if there is a plotting tool that visualizes a local range of the genome (e.g. a window big enough to inspect a gene) that integrates (in one plot):
1. Point mutations in the region
2. The local copy numbers (major/minor allele if available)
3. SVs affecting the region
Note that the purpose is more to infer the consequence of a mutation as opposed to validating whether the mutation is real or not, which we can already do with a variety tools such as samtools tview.
Circos is a nice format that integrates all three classes of mutations, but it is more for global scale inspections and does not provide enough resolution to zoom into specific gene regions in order to answer the above questions about key mutations of interest. Another close one is IGV, but it does not incorporate allelic specific copy number info.
Thanks! | You can give a try to [karyoploteR](http://bioconductor.org/packages/karyoploteR/). It's an R package to plot data on genomes and can go from single base to the whole genome. Take a look at the [tutorial and examples](https://bernatgel.github.io/karyoploter_tutorial/) to see what it looks like. It's only a plotting tool and does not know much biology, so it can plot almost anything as long as you have it precomputed and can map it to any of the available plot types. Right now it does not have the possibility of plotting individual sequencing reads from a BAM file, if you need that.
A couple of examples of whole genome views.
Note: Setting `zoom` to a small genomic region in the call to `plotKaryotype` will created a zoomed in version of the plots with the exact same code.
![enter image description here][1]
![enter image description here][2]
![enter image description here][3]
[1]: https://bernatgel.github.io/karyoploter_tutorial/Examples/SNPArray/images/Figure4-1.png
[2]: https://bernatgel.github.io/karyoploter_tutorial/Examples/GeneExpression/images/Figure13-1.png
[3]: https://bernatgel.github.io/karyoploter_tutorial/Examples/Rainfall/images/Figure5-1.png | biostars | {"uid": 350697, "view_count": 2559, "vote_count": 5} |
I have a SAM file with alignments and for each entry alignment, I want to reconstruct the alignment between the reference and the read based on the CIGAR and MD strings. It seems like this should be possible, but this one example bothers me:
SRR037452.3355 0 ENSG00000266658|ENST00000607521 3523 255 16M1I18M * 0 0 CGGGCCGGTCCCCCCCCGCCGGGTCCGCCCCCGGC IIIIIIIIIIIIIII:III/IIII=+IGC,I"/I. NH:i:1 HI:i:1 NM:i:2 MD:Z:33G0
Here, the CIGAR string has an insertion to the reference which messes up the MD string indexing. According to MD string, the read should be only 34 bases long (r=35). My guess is that the alignment is actually this: 16 matches, 1 base that is present in the read, not present in the reference, 17 matches, one mismatch where the read has a "G" and the reference has something else. Is that correct? Are there CIGAR/MD string combinations where reconstructing the alignment would be impossible (i.e. either of the strings is ambiguous)? | <p>Your guess is correct, the alignment has 16 matches, 1 unspecified insertion, followed by 17 matches and one mismatch ('G' on the reference which is substituted with a 'C' in the read sequence).</p>
<p>If the MD tag is present, you should always be able to reconstruct the alignment from the MD tag and the CIGAR, as long as you don't care about the sequence of any mismatched or inserted bases (which you can only get from the read sequence).</p>
| biostars | {"uid": 112382, "view_count": 7962, "vote_count": 7} |
Hello, I have phased plink files of a population. I would like to obtain all SNPs that are in high linkage disequilibrium (LD) for a set of 10 SNPs. I know how to do it according to a r2 threshold, but I need to set this threshold according to D'
For instance, this script will output all variants in high LD (r2 > 0.6) for a list of 10 SNPs within a range of 1000 Kb.
plink \
--bfile myfile \
--r2 --ld-snp-list list_10_snps.txt \
--ld-window-kb 1000 --ld-window-r2 0.6
Thanks very much | This isn't directly supported by plink, but you can use "--r2 dprime" to include D' in the main LD report, and then follow up with e.g. an awk one-liner to filter on D'. | biostars | {"uid": 360371, "view_count": 2026, "vote_count": 2} |
How to embed known cell information into Seurat object? Using AddMetaData?
Is this possible?
I read this documentation: https://www.rdocumentation.org/packages/Seurat/versions/3.1.4/topics/AddMetaData
However I need some clarification on how to go about doing this, please?
I have cell labels that are all aligned with the expression matrix such as this:
"12wks_fetal pancreas cell_acinar" "19wks_fetal pancreas cell_beta" "12wks_fetal pancreas cell_ductal" "12wks_fetal pancreas cell_beta" "12wks_fetal pancreas cell_acinar" "22wks_fetal pancreas cell_acinar" "12wks_fetal pancreas cell_acinar" "12wks_fetal pancreas cell_alpha" "12wks_fetal pancreas cell_ductal" "12wks_fetal pancreas cell_acinar" "14wks_fetal pancreas cell_endocrine.progenitor..."
I used these cell labels as cell names (column names) for the expression matrix. However that's not useful for seeing which cell is from what time point 12wks, 19wks, 14 wks, etc. on the UMAP for example.
Would there be a way to label the cells using the AddMetaData function so that when I visualize the data using UMAP, they are color coded based on the time of collection (ie. 12wks, 19wks, 22wks, etc.) or would there be a way I could also filter by both time of collection and/or cell type?
Is this possible in Seurat? How would I approach this?
I would really appreciate anyone's help.
Very Respectfully,
Pratik
| You didn't think to try AddMetaData?
Yes, if you have one column with barcode names an another with any kind of information, you can add a new column of metadata. You can also make a new column of metadata by concatenating two columns you already have. You can use group.by in DimPlot to color the cells by any column in the metadata, and you can subset by metadata columns after setting that column with Ident. | biostars | {"uid": 460935, "view_count": 24192, "vote_count": 1} |
Probably a basic question but it's difficult to find information on the subject...
When viewing a GTF and BED file in IGV, there seems to be differences in colour (BED track can't be recoloured, just shows as black, GTF can be recoloured). Does anyone know the reason for this? I thought that the GTF and BED formats contained predominantly the same information just in different formats.
Thanks! | <p>A GTF file is more akin to a large group of BED files grouped according to some feature or features of relevance, often with multiple levels of hierarchical metadata. In the classical case of genes, GTF specifies a standard way of relating exons to transcripts and genes. There's no standard way of doing that with BED files. BED files are useful for cases where the file itself contains only a single type of information (e.g. ChIP-seq peaks), where you could instead combine multiple of these files in a GFF (e.g., annotating peaks from different histones pulled down with multiple antibodies each). You can do some of this by using the `name` field in the BED format, but there's still no good way to represent hierarchical.</p>
| biostars | {"uid": 97868, "view_count": 4222, "vote_count": 2} |
I have been using an HPC cluster for a few years now and regularly need to submit jobs that process large amounts (often over 100) for large files like BAM files etc.
Despite some experience, I feel I am lacking some of the understanding of the basic concepts such as:
- How to estimate how much RAM and runtime a job will need - I know, it's mostly based on experience and no one can ever answer that for me
- The relationship between how much RAM you give a job and how much runtime? Are these 2 parameters independent? Will one affect how long you are in the queue more?
**So my question is:**
Does anyone know of a nice book/online resource that explains these basic concepts and ideas? I find myself struggling to answer these simple questions and the documentation out there is very often geared towards explaining complicated details about how supercomputers work. I am interested in all that but I would like to start with a dummed-down version first that focusses on how to submit jobs properly.
Any ideas? I should say, the cluster I use works with the Sun Grid Engine[SGE] system.
| 1. Nope, you just guess, then optimize.
2. Nope, not usually. I have a few very lazy scripts that use a ton of RAM (I have it, why not?) they're fast, some things use very little ram and can take a relatively long time.
So, if your goal was to come up with some metric to allocate your job wall time limits on the RAM for the job, don't, it won't work.
> I find myself struggling to answer these simple questions and the documentation out there is very often geared towards explaining complicated details about how supercomputers work.
I think you sort of answered it yourself, you really only need to dig into that documentation if you're doing things that really rely on the intricate details of a cluster. Just like with coding, don't worry about optimizing till you need to.
Here are some tips:
- Don't worry about over allocation of resources. As long as you're not
allocating 50 128 core/1TB RAM nodes to run 1 instance of gzip each,
you'll be fine. If you take a guess that a job needs 64GB and you
only use 47, its not the end of the world. If you ask for 8 cores and
only manage to keep 6 loaded, oh well, next time use less.
- Considering most systems kill jobs that exceed their allocated
resources, it doesn't hurt to round up. If you undershoot and the job
gets killed, you've still wasted wall time. "Wasting" some ram and
some cores is better than restarting an 8 hour job because you needed
64 and only asked for 48.
- Over-allocating wall time is even less of an issue, the resources are
free once the job is done, if it takes 1 hour or 1 month. All that
happens is your jobs may have less priority over shorter ones.
- Typically programs that rely on a database need that much memory to
load the database. So, unless you know the DB stays on the disk, a
good idea is adding the size of the query and database and
multiplying by 1.25. Tweak accordingly.
- Small, short jobs take priority over long, large jobs.
- Don't constantly undershoot your wall time, you'll either piss off
the admins with constant requests for increases or constantly waste
time having to resubmit.
- Make good use of the scratch space on nodes and be careful about
bogging down the file system by having a ton of jobs trying to I/O
the same files or directories. If jobs need to constantly access
something (say an index for HiSat2), copy it to the scratch, and have
the job read it from there.
- Interactive sessions are great. Use them to debug and benchmark jobs.
This is a great tool to find out little things that get you in
trouble, like a program starting more threads than the CPUs you
allocated for it.
- Different programs behave differently as you add cores. Some don't
have substantial memory increases, some do. Experiment to find out.
- When in doubt, ask the admins. They know the system and may have
helped another user do something similar.
Try stuff, break stuff, bother the admins. Your goal is to get work done, the cluster lets you do if faster, you're not there to optimize HPC software. A cluster is a tool, use it like a hammer. | biostars | {"uid": 335623, "view_count": 2293, "vote_count": 3} |