# Oligomerization of Sulfolobus solfataricus signature amidase is promoted by acidic $\mathrm{pH}$ and high temperature
ANNA SCOTTO D'ABUSCO, ${ }^{1}$ RITA CASADIO, ${ }^{2}$ GIANLUCA TASCO, ${ }^{2}$ LAURA
GIANGIACOMO, ${ }^{1}$ ANNA GIARTOSIO, ${ }^{1}$ VALENTINA CALAMIA, ${ }^{1}$ STEFANIA DI MARCO, ${ }^{3}$
ROBERTA CHIARALUCE, ${ }^{1}$ VALERIO CONSALVI, ${ }^{1}$ ROBERTO SCANDURRA ${ }^{1,4}$ and LAURA
POLITI ${ }^{1}$
${ }^{1}$ Dipartimento di Scienze Biochimiche "A. Rossi-Fanelli," Università "La Sapienza," P.le A. Moro 5, 00185 Roma, Italy
${ }^{2}$ CIRB Biocomputing Group, University of Bologna, Via Irnerio 42, 40126 Bologna, Italy
${ }^{3}$ IRBM P. Angeletti, Via Pontina Km 30.600, 00040 Pomezia, Roma, Italy
${ }^{4}$ Corresponding author (roberto.scandurra@ uniromal.it)
Received July 4, 2005; accepted September 13, 2005; published online October 21, 2005
#### Abstract
Summary The recombinant amidase from the hyperthermophylic archaeon Sulfolobus solfataricus (SSAM) a signature amidase, was cloned, purified and characterized. The enzyme is active on a large number of aliphatic and aromatic amides over the temperature range $60-95^{\circ} \mathrm{C}$ and at $\mathrm{pH}$ values between 4.0 and 9.5 , with an optimum at $\mathrm{pH} 5.0$. The recombinant enzyme is in the form of a dimer of about $110 \mathrm{kD}$ that reversibly associates into an octamer in a $\mathrm{pH}$-dependent reaction. The $\mathrm{pH}$ dependence of the state of association was studied using gel permeation chromatography, analytical ultracentrifugation and dynamic light scattering techniques.
At $\mathrm{pH} 7.0$ all three techniques show the presence of two species, in about equal amounts, which is compatible with the existence of a dimeric and an octameric form. In decreasing $\mathrm{pH}$, the dimers formed the octameric species and in increasing $\mathrm{pH}$, the octameric species was converted to dimers. Above $\mathrm{pH} 8.0$, only dimers were present, below $\mathrm{pH} 3.0$ only octamers were present. The association of dimers into octamers decreased in non-polar solvents and increased with temperature. A mutant (Y41C) was obtained that did not show this behavior.
Keywords: amidase signature, amide metabolism, hyperthermophile.
## Introduction
Amidases, a large class of enzymes broadly spread over all three domains of living organisms, act on $\mathrm{C}-\mathrm{N}$ bonds other than peptide bonds and catalyze the hydrolysis of various endogenous and foreign aliphatic and aromatic amides by transferring an acyl group to water producing free acids and ammonia. The discovery of an amidase signature gene family (Mayaux et al. 1991) allowed grouping of the primary structure of the codified enzymes into two classes, the first sharing a characteristic signature and the second lacking this signature. Amidase signature family enzymes include at least 200 proteins (most of these are putative, being derived from DNA data) from 90 different organisms distributed among Bacteria, Archaea and Eukarya (Altschul et al. 1997). These enzymes are characterized by the presence of the sequence GGSS(S/G)GS in a conserved stretch of about 130 amino acids (the "amidase signature sequence") in the center of the protein (Mayaux et al. 1991, Chebrou et al. 1996). The biological functions of the amidase signature family enzymes include the formation of Gln-tRNA ${ }^{\mathrm{GIn}}$ through the transamidation of misacylated Glu-tRNA ${ }^{\mathrm{Gln}}$ by amidolysis of glutamine in many bacteria (Curnow et al. 1997, Schon et al. 1998), the formation of indole-3-acetic acid in pathogenic plant bacteria (Gaffney et al. 1990), the metabolic turnover of foreign and endogenous amides in prokaryotes and eukaryotes (Mayaux et al. 1991, Gomi et al. 1991) and the catabolism of neuromodulatory fatty acid amides in mammals (Koutek et al. 1994, Cravatt et al. 1996, Patricelli and Cravatt 2000).
In a previous study, we cloned and expressed the signature amidase from the archaeon Sulfolobus solfataricus MT4 as a fusion protein with a His-tag at the N-terminus (Scotto d'Abusco et al. 2001). Preliminary experiments on samples enzymatically deprived of the His-tag showed that the protein may undergo association-dissociation processes induced by $\mathrm{pH}$. To further investigate this finding, we expressed the recombinant protein without the tag. The association-dissociation processes of the protein were investigated by gel permeation chromatography (GPC), analytical ultracentrifugation (AUC) and dynamic light scattering (DLS), and the results are presented in this paper. Random mutagenesis was performed followed by selection for mutants that did not undergo $\mathrm{pH}$-induced association. Among the mutants obtained, the mutant Y41C, which has a fifth cysteine in addition to the four that
form the two disulfide bridges in the wild type (wt) protein, attracted our attention. Unlike the wt protein, the Y41C mutant did not exhibit the reversible $\mathrm{pH}$-dependent association.
The 3D structure of $S$. solfataricus amidase (SSAM) is not available; therefore, we computed a low-resolution model of the protein based on the peptide amidase (PAM) from Stenotrophomonas maltophila (Labahn et al. 2002).
## Materials and methods
## Protein expression and purification
Amidase expression was obtained by amplifying the amidase gene through PCR and inserting the PCR product into $\mathrm{pET3d}$ expression vector (Invitrogen, Carlsbad, CA). The clone $\lambda \mathrm{C}$, previously described (Scotto d'Abusco et al. 2001), was used as the template.
The PCR reaction was performed with the primers AMS (5'GTCCATGGGAATTAAGTTACCCA3') and AMS (5' GAGAGGATCCTTATTTTTTGATTCT ${ }^{\prime}$ ) with the following conditions: $94^{\circ} \mathrm{C}$ for $1 \mathrm{~min} ; 56^{\circ} \mathrm{C}$ for $1 \mathrm{~min}$; and $72^{\circ} \mathrm{C}$ for 2 min for 30 cycles. The PCR product was digested with Nco I and Bam $\mathrm{HI}$ and inserted into $\mathrm{pET3d}$, digested with the same pair of enzymes. The construct obtained, pASC/2E, confirmed by DNA sequencing, was transformed into E. coli BL21 (DE3)pLysS strain (Invitrogen, Carlsbad, CA) for protein expression. Cells were grown at $37^{\circ} \mathrm{C}$ in Luria-Bertani (LB) medium with ampicillin ( $100 \mu \mathrm{g} \mathrm{ml}{ }^{-1}$ ) and $0.4 \mathrm{mM}$ of isopropyl- $\beta$-D-thiogalactopyranoside (IPTG) was added when the culture reached $1.6 \mathrm{OD}_{600}$. Cells were harvested by centrifugation and disrupted by sonication. The recombinant enzyme was found in the insoluble fraction, which was extracted three times with $20 \mathrm{mM}$ sodium phosphate buffer, $\mathrm{pH} 7.8$, containing $500 \mathrm{mM} \mathrm{NaCl}$ and $0.25 \%$ Tween 20. Samples containing $5 \mathrm{mg}$ of protein were applied on a Hiload 16/60 Superdex 200 column (Amersham Biosciences Europe) and eluted with $20 \mathrm{mM}$ sodium phosphate buffer, $\mathrm{pH} 7.0$, containing $150 \mathrm{mM} \mathrm{NaCl}$ at $1 \mathrm{ml} \mathrm{min}^{-1}$. The purified protein was concentrated with an Amicon pressurized cell equipped with a PM 30 membrane (Amicon, Beverly, MA). The purity of the protein was $98 \%$ as checked through gel electrophoresis performed either under native or denaturing conditions.
## Random mutagenesis
The mutagenesis reaction was performed with the Diversify ${ }^{\mathrm{TM}}$ Random Mutagenesis Kit (Clontech, Palo Alto, CA) according to the instruction manual. The conditions for PCR mutagenesis were optimized to produce an average of two base pair substitutions per molecule within the amidase gene (wt) from S. solfataricus. The template was pASC/2E. The forward primer was -T7 (5'-TAATACGACTCACTATAGGG-3'), and the reverse primer was -T7R (5'-ATCAATAACGAGTCGCC $\mathrm{ACC}-3^{\prime}$ ). The PCR reaction was carried out at $94^{\circ} \mathrm{C}$ for $30 \mathrm{~s}$; $46^{\circ} \mathrm{C}$ for $30 \mathrm{~s}$; and $72^{\circ} \mathrm{C}$ for $1.5 \mathrm{~min}$ for 25 cycles. The PCR reaction was checked by agarose gel and the amplified DNA fragments were purified with a Nucleospin extract Kit (Macherey-Nagel, Düren, Germany). The amplified DNA fragments and pET3d were digested separately by Nco I and Bam HI restriction enzymes and ligated. The resulting plasmid was used to transform E. coli BL21 (DE3)pLysS strain. Five clones were analysed by sequencing. The expression and purification of the mutant protein were performed as reported above for the wt recombinant protein.
## Protein determination
Protein concentration was determined at $280 \mathrm{~nm}$ based on an extinction coefficient of $54,290 \mathrm{M}^{-1} \mathrm{~cm}^{-1}$, calculated according to Gill and von Hippel (1989) as reported by Scotto d'Abusco et al. (2001).
## Enzyme assay
The standard enzyme assay was performed at $70^{\circ} \mathrm{C}$ for 10 min with $7.5 \mathrm{mM}$ benzamide in $50 \mathrm{mM}$ citrate buffer at $\mathrm{pH} 5.0$. The amount of benzoic acid produced was determined by high performance liquid chromatography (Scotto d'Abusco et al. 2001). The $\mathrm{pH}$ of buffers was checked at the assay temperatures.
## The $\mathrm{pH}$ optimum
The $\mathrm{pH}$ optimum for the amidase reaction was determined at $70^{\circ} \mathrm{C}$ for $10 \mathrm{~min}$, using the following buffers: citrate buffer for $\mathrm{pH} 3.0-5.8$, sodium phosphate buffer for $\mathrm{pH} 6.0-7.9$ and BIS-TRIS propane buffer for $\mathrm{pH} 6.5-9.5$. The concentration of all buffers was $50 \mathrm{mM}$ and their $\mathrm{pH}$ was checked at the assay temperature. The substrate was $7.5 \mathrm{mM}$ benzamide.
## Biochemical characterizations
The kinetic constants with substrates listed in Table 1 were determined at $70^{\circ} \mathrm{C}$ in $200 \mu$ of $50 \mathrm{mM}$ citrate buffer at $\mathrm{pH} 5.0$, by measuring the free acid produced by HPLC, as described by Scotto d'Abusco et al. (2001). Kinetic data were calculated by nonlinear regression analysis using ENZFITTER (Leatherbarrow 1990). For each calculation, there were at least seven velocity-substrate data pairs.
## Chemical modifications
Arginine residues were reacted with phenylglyossal as reported elsewhere (Riordan and Scandurra 1972) and arginine modification was deduced with an amino acid analyzer after hydrolysis of the protein in $6 \mathrm{~N} \mathrm{HCl}$ at $110^{\circ} \mathrm{C}$ in vacuo for $22 \mathrm{~h}$.
## Gel permeation chromatography
Protein samples were incubated overnight at room temperature in $50 \mathrm{mM}$ citrate buffer for $\mathrm{pH} 3.0-5.0$, in sodium phosphate for $\mathrm{pH}$ 6.0-7.0 and BIS-TRIS propane buffer for $\mathrm{pH} 8.0-9.0$ and then injected on a Superdex 200 column (Amersham Biosciences Europe) equilibrated with $20 \mathrm{mM}$ sodium phosphate buffer, $\mathrm{pH} 7.0$, containing $0.15 \mathrm{M} \mathrm{NaCl}$. The protein was eluted at room temperature in the same buffer at $0.4 \mathrm{ml} \mathrm{min}^{-1}$. The eluate was monitored at 226 and $280 \mathrm{~nm}$. The column was calibrated with the following standards (Amersham Biosciences Europe): thyroglobulin ( $669 \mathrm{kDa}$ ),
Table 1. Kinetic properties of Sulfolobus solfataricus amidase and its Y41C mutant (in bold). The turnover number $\left(k_{\text {cat }}\right)$ was obtained dividing by the molecular mass of the active monomer (55,784 Da); temperature was $70^{\circ} \mathrm{C}$; there were $1-10 \mu \mathrm{g}$ of enzyme in a total volume of $200 \mu \mathrm{l}$ incubated for $2-10 \mathrm{~min}$ in $50 \mathrm{mM}$ citrate buffer at $\mathrm{pH}$ 5.0. Abbreviation: $K_{\mathrm{m}}=$ Michaelis constant.
| Substrate | $K_{\mathrm{m}}$
$(\mathrm{mM})$ | $k_{\mathrm{cat}}$
$\left(\mathrm{sec}^{-1}\right)$ | $k_{\mathrm{cat}} / K_{\mathrm{m}}$
$\left(\mathrm{M}^{-1} \sec ^{-1}\right) \times 10^{2}$ |
| :---: | :---: | :---: | :---: |
| Acetamide | 22.0 | 28.9 | 13.1 |
| | 37.0 | 30.1 | 8.1 |
| Propionamide | 10.0 | 82.9 | 82.9 |
| | $\mathbf{1 0 . 0}$ | 99.4 | 99.4 |
| Butyramide | 14.0 | 34.3 | 24.5 |
| | 3.2 | 17.5 | 54.7 |
| Isobutyramide | 7.0 | 48.9 | 69.9 |
| | 6.5 | 58.4 | 89.8 |
| Pentanamide | 2.5 | 26.8 | 107.2 |
| | 1.5 | 28.6 | 190.6 |
| Hexanamide | 4.5 | 19.5 | 43.3 |
| | 4.5 | 21.6 | 48.0 |
| Metacrylamide | 0.3 | 4.8 | 160.0 |
| | 0.5 | 3.0 | 60.0 |
| Benzamide | 0.9 | 6.7 | 74.4 |
| | 0.6 | 7.2 | 120.0 |
| p-OH benzamide | 0.1 | 1.9 | 190.0 |
| | 0.1 | 2.1 | 210.0 |
| p-NH2 $\mathrm{NH}_{2}$ enzamide | 0.2 | 0.8 | 40.0 |
| | 0.3 | 2.5 | 83.3 |
| o-toluamide | 0.1 | 0.4 | 40.0 |
| | 0.2 | 0.8 | 40.0 |
| p-toluamide | 0.2 | 12.2 | 610.0 |
| | 0.3 | 14.2 | 473.3 |
| Nicotinamide | 1.5 | 6.4 | 42.6 |
| | 0.4 | 2.2 | 55.0 |
| 3-phenylpropionamide | 3.0 | 15.3 | 51.0 |
| | 2.5 | 18.0 | 72.0 |
| Indol-3-acetamide | 0.8 | 2.2 | 27.5 |
| | 0.8 | 1.2 | 15.0 |
ferritin (440 kDa), catalase ( $232 \mathrm{kDa}$ ), aldolase ( $158 \mathrm{kDa}$ ), albumin ( $67 \mathrm{kDa}$ ), ovoalbumin ( $43 \mathrm{kDa}$ ) and ribonuclease ( $13.7 \mathrm{kDa}$ ). The molecular mass of amidase was calculated by plotting the log of molecular mass versus elution volume (Ve) using a linear regression curve of the Unicorn 4.10 package for control and evaluation data of the Akta Chromatograph from Amersham Biosciences
## Analytical ultracentrifugation
Sedimentation velocity experiments were conducted at $1.17 \times 10^{5} \mathrm{~g}$ and $20^{\circ} \mathrm{C}$ in a Beckman Optima XL-A analytical ultracentrifuge equipped with absorbance optics and an An60Ti rotor at a protein concentration of $0.5 \mathrm{mg} \mathrm{m1} 1^{-1}$. The buffer used was $50 \mathrm{mM}$ sodium phosphate at $\mathrm{pH} 6.0$ and 8.0. Equilibration with the desired buffer was achieved through dialysis or by passage through a Sephadex G25 column. Data were collected at $278 \mathrm{~nm}$ at a spacing of $0.005 \mathrm{~cm}$ with three averages in a continuous scan mode every 4 min and analyzed with the program Sedfit (Schuck and Rossmanith 2000), provided by Dr. Peter Schuck (NIH, Bethesda, MD).
## Dynamic light scattering
Dynamic light scattering measurements were performed with a DynaPro-801 instrument with temperature control (Protein Solution, Charlottsville, VA), where the scattered light was collected at an angle of $90^{\circ}$ through a fiber optic and converted to an electrical signal by an avalanche photodiode. The timedependent autocorrelation function of the photon current was monitored with a 20 -channel software correlator (based on a Digital Signal Processor unit) provided by the manufacturer The first sampling time was $3.84 \mu \mathrm{s}$. The length of the subsequent channels increases in a quasi-logarithmic fashion. The samples were gently injected into the cell through a series of Whatman filters with decreasing porosity, from 0.22 to $0.02 \mu \mathrm{m}$. Protein concentrations for both the wt amidase and the Y41C mutant ranged from 3.0 to $0.2 \mathrm{mg} \mathrm{ml}^{-1}$. After an overnight incubation in the appropriate buffer, measurements were performed first at $3.0 \mathrm{mg} \mathrm{ml}^{-1}$ and then, after dilution with the same buffer, repeated at $2.6,1.3,0.5,0.3$ and $0.2 \mathrm{mg}$ $\mathrm{ml}^{-1}$. Buffers used for measurements were $20 \mathrm{mM}$ sodium phosphate at $\mathrm{pH} 8.0$ and $20 \mathrm{mM}$ sodium citrate at $\mathrm{pH} 4.0$. Autocorrelation functions were measured every $60 \mathrm{~s}$, based on $0.18 \times 10^{5}$ to $3.3 \times 10^{5}$ counts each. Data analyses were performed with Protein Solutions' Dynamics analysis software. Measurements were made at $20.1 \pm 0.2^{\circ} \mathrm{C}$.
## Spectroscopic analyses
Intrinsic fluorescence emission measurements were made with a Perkin-Elmer LS50B spectrofluorimeter with a $1 \mathrm{~cm}$ pathlength quarz cuvette. Fluorescence emission spectra were recorded between 300 and $450 \mathrm{~nm}$ ( $1 \mathrm{~nm}$ sampling interval) at $20^{\circ} \mathrm{C}$ with the excitation wavelength set at $295 \mathrm{~nm}$. All the spectra were recorded at $0.1 \mathrm{mg} \mathrm{ml}^{-1}$ protein concentration after an overnight incubation in $20 \mathrm{mM}$ sodium phosphate buffer at $\mathrm{pH} 8.0$ or $20 \mathrm{mM}$ sodium citrate buffer at $\mathrm{pH} 5.0$. Experiments with the fluorescent dye 8 -anilinonaphthalene-1-sulfonic acid ammonium salt (ANS) were performed at $20^{\circ} \mathrm{C}$ by incubating the protein and ANS at a 1:10 molar ratio. After $5 \mathrm{~min}$, fluorescence emission spectra were recorded at 400 $600 \mathrm{~nm}$ with the excitation wavelength set at $390 \mathrm{~nm}$. The CD spectra were recorded on a Jasco J-720 spectropolarimeter The results are expressed as mean residue ellipticity assuming a mean residue of 110 per amino acid residue.
## Monomer detection
The dissociation of the dimer was attempted by adding either urea (up to $2 \mathrm{M}$ ) or guanidinium chloride (up to $0.8 \mathrm{M}$ ), with thiocyanate (up to $0.5 \mathrm{M}$ ) to a solution of wt protein or $\mathrm{Y} 41 \mathrm{C}$ mutant or by increasing the $\mathrm{pH}$ to 11.0. Protein samples in $50 \mathrm{mM}$ BIS-TRIS propane buffer $\mathrm{pH} 8.0$ (where only dimers are present) were incubated for $2 \mathrm{~h}$ at $20^{\circ} \mathrm{C}$ with the mentioned compounds and injected in a GPC column.
## Thermoactivity
The temperature dependence of the amidase reaction was studied under standard conditions over the range $40-100^{\circ} \mathrm{C}$.
## Thermal stability
The measurements were conducted at 70,80 and $90^{\circ} \mathrm{C}$ in Eppendorf tubes $\left(0.2 \mathrm{ml}\right.$ ) using $0.2 \mathrm{mg} \mathrm{ml}^{-1}$ protein in $70 \mu \mathrm{l}$ $50 \mathrm{mM}$ citrate buffer at $\mathrm{pH} 5.0$. The tubes were sealed and incubated for the reported times in a Thermomixer compact (Eppendorf, Hamburg, Germany). After incubation a $50 \mu \mathrm{l}$ sample was assayed for standard activity.
## Differential scanning calorimetry
Heat capacity versus temperature profiles were obtained with a VP-DSC differential scanning calorimeter (MicroCal.Inc., Northampton, MA) at a heating rate of $60^{\circ} \mathrm{Ch}^{-1}$. Protein solutions ( $0.1-0.3 \mathrm{mg} \mathrm{ml}^{-1}$ ) in the appropriate buffer were degassed at $25{ }^{\circ} \mathrm{C}$ before the calorimetric experiments. The reference cell was filled with the same solvent mixture used for the sample, but lacking the protein. Both cells were kept under an excess pressure of $200 \mathrm{~Pa}$ to avoid bubbling during the scan. At the end of the run the solutions were subjected to a second heating cycle under the same conditions to determine the reversibility of the transitions. Thermograms were corrected by subtracting the instrumental baseline, obtained with both cells filled with the same solvent, and normalized for protein concentration. The $T_{\mathrm{m}}$ (temperature at which excess heat capacity reaches a maximum, ${ }^{\circ} \mathrm{C}$ ) was determined with the ORIGIN software provided by MicroCal, after subtraction of a cubic baseline connecting the pre- and post-transition traces.
## Construction of the model
Three signature amidases with different metabolic roles are presently known at the atomic resolution: malonamidase E2 (MAE2) from Bradyrhizobium japonicum (Shin et al. 2003), the fatty acid amide hydrolase from Rattus norvegicus (FAAH) (Bracey et al. 2002) and the peptide amidase (PAM) from Stenotrophomonas maltophila (Labahn et al. 2002). The SSAM 3D model was built with PAM as a template. This choice was suggested by a better sequence alignment of the target/template (complete coverage, identity level $=30 \%$ ) compared with the other two sequences and also because SSAM and PAM use similar substrates. The 3D structure was computed after template/target alignment with Modeller v. 6.2 (Sali and Blundell 1993). After prediction of the interaction patches with ISPRED (Fariselli et al. 2002), the dimer form was built with the ZDOCK tool (Chen and Weng 2002).
## Results
The expression of recombinant wt $S$. solfataricus amidase by the pET3d expression vector, allowed us to obtain the enzyme in its native form. Furthermore, random mutagenesis was applied in order to find mutants showing physico-chemical properties or substrate specificities or both that are different from those found in the recombinant $S$. solfataricus wt amidase. Mutant amidases from all the clones obtained were expressed and purified and their physico-chemical properties analyzed. Mutant amidase with the Y41C substitution was selected because it behaved differently from the wt protein as a function of $\mathrm{pH}$.
## pH optimum and kinetic parameters
As reported in Figure 1, the activities of recombinant wt amidase and its mutant, measured at $70^{\circ} \mathrm{C}$, showed $\mathrm{pH}$ optima at $\mathrm{pH}$ 5.0. Therefore this $\mathrm{pH}$ was chosen for measurements of kinetic parameters on a number of aromatic and aliphatic amides (Table 1).
## Gel permeation chromatography
After overnight incubation at room temperature of the wt protein in various $50 \mathrm{mM}$ buffers at various $\mathrm{pHs}$, the samples were injected onto a Superdex 200 column eluted at $0.4 \mathrm{ml} \mathrm{min}^{-1}$ with $20 \mathrm{mM}$ sodium phosphate buffer at $\mathrm{pH} 7.0$ and $0.15 \mathrm{M}$ $\mathrm{NaCl}$. The elution pattern of a sample incubated in $50 \mathrm{mM}$ BIS-TRIS propane buffer $\mathrm{pH} 7.2$ is shown in Figure 2A. Two peaks with elution volumes corresponding to a molecular mass of about $83 \mathrm{kDa}$ and $400 \mathrm{kDa}$, compatible with a dimeric (D) and octameric $(\mathrm{O})$ species, respectively, were obtained. The ratio of the two species was dependent on $\mathrm{pH}$. The distribution of the area of the two peaks as a function of $\mathrm{pH}$ is shown in the inset of Figure 2A: a decrease in $\mathrm{pH}$ resulted in increasing formation of the oligomeric species (open circles) and a parallel reduction of the dimeric species (open squares). Conversely, the latter species increased with increasing $\mathrm{pH}$ : from $\mathrm{pH} 8.0$
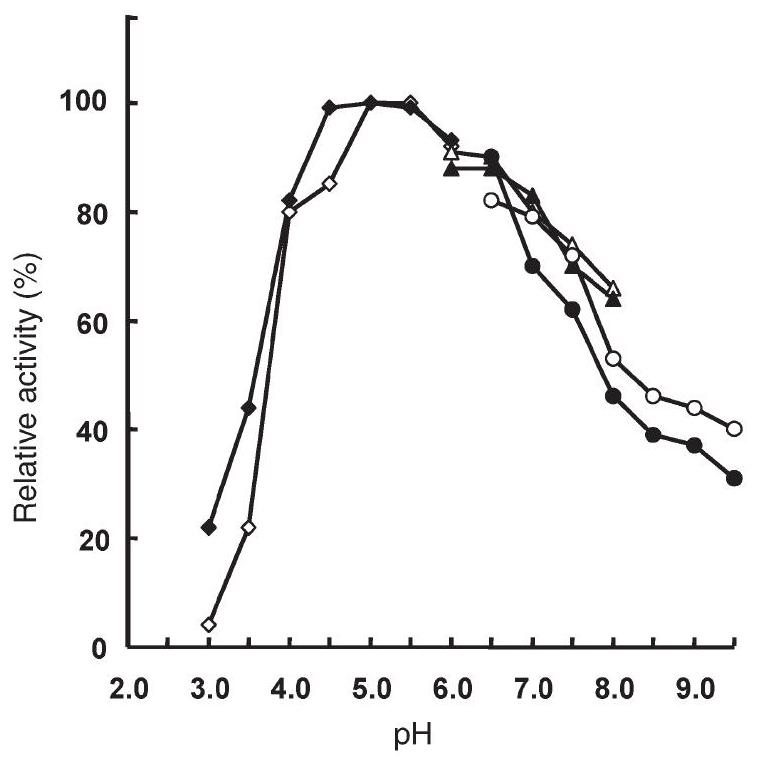
Figure1. Effect of $\mathrm{pH}$ on the activity of Solfolobus solfataricus recombinant wild type (wt) amidase (filled symbols) and its Y41C mutant (open symbols). Enzyme activity was measured at $70^{\circ} \mathrm{C}$ for $10 \mathrm{~min}$, with $7.5 \mathrm{mM}$ benzamide in $50 \mathrm{mM}$ citrate buffer at $\mathrm{pH} 4.0-5.8(\bullet$, $\diamond$ ), sodium phosphate buffer at $\mathrm{pH} 6.0-7.9(\mathbf{\Delta}, \triangle)$ and BIS-TRIS propane buffer at $\mathrm{pH} 6.5-9.5(\bullet, O)$. The $\mathrm{pH}$ of each buffer was checked at $70^{\circ} \mathrm{C}$. The $100 \%$ activity corresponds to 7.2 and 7.8 units $\mathrm{mg}^{-1}$ of wt and mutant proteins, respectively ( 1 enzyme unit $=1 \mu \mathrm{mol}$ of free acid formed per minute).
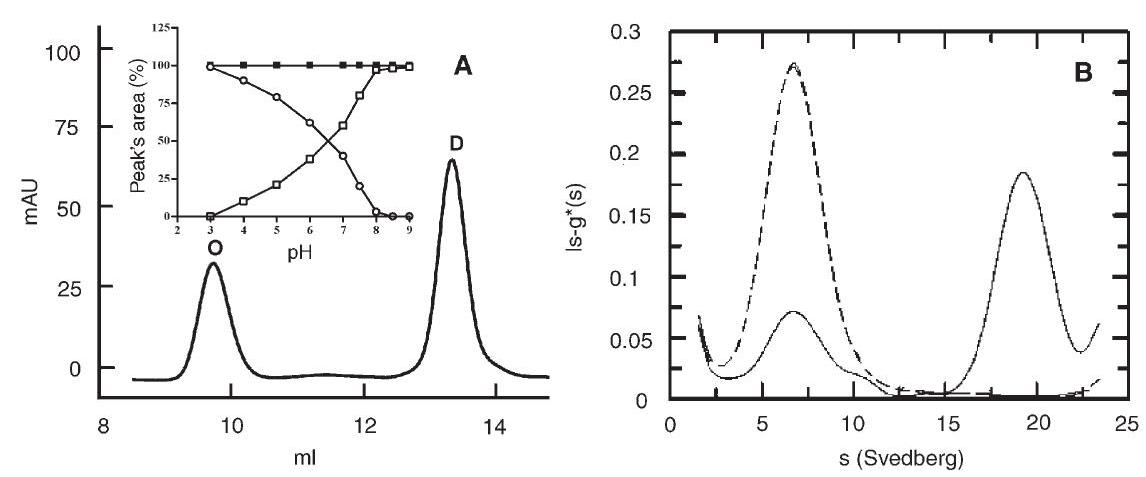
Figure 2. Effect of $\mathrm{pH}$ on the oligomeric state. (A) Gel permeation chromatography: $30 \mu \mathrm{g}$ of protein was incubated overnight in $50 \mathrm{mM}$ BIS-TRIS propane buffer at $\mathrm{pH} 7.2$, then injected onto the Superdex 200 column, eluted at room temperature at $0.4 \mathrm{ml} \mathrm{min}^{-1}$ with $20 \mathrm{mM}$ phosphate buffer containing $150 \mathrm{mM} \mathrm{NaCl}$ and monitored at $226 \mathrm{~nm}$. Abbeviations: $\mathrm{O}=$ octamer; $\mathrm{D}=$ dimer. The inset shows the percentage of each peak's area from the elution patterns of wild type (wt) protein samples obtained after overnight incubation with buffers at various $\mathrm{pH}$ values (open circles $=$ octamer; open squares $=$ dimer). The filled squares show the percentage of the dimer's area obtained with Y41C mutant samples after overnight incubation with buffers at various $\mathrm{pH}$ values. (B) Analytical ultracentrifugation: the wt protein concentration was $0.5 \mathrm{mg} \mathrm{ml}^{-1}$ in $50 \mathrm{mM}$ phosphate buffer at $\mathrm{pH} 6.0$ (solid line) and 8.0 (dashed line). Experiments were conducted at $1.17 \times 10^{5} \mathrm{~g}$ and $20^{\circ} \mathrm{C}$, at a wavelength of $278 \mathrm{~nm}$ after reaching equilibrium ( $12 \mathrm{~h}$ ). Peak ( $80 \%$ ) with $\mathrm{S}_{\mathrm{w}, 20}=18$ corresponds to the octamer (about $400 \mathrm{kDa}$ ). Peak (20\%) with $\mathrm{S}_{\mathrm{w}, 20}=6$ corresponds to the dimer (about $100 \mathrm{kDa}$ ).
upward, only the dimeric species was present, whereas below $\mathrm{pH} 3.0$, only the octameric species was present. Conversion of one species of the wt protein to the other induced by $\mathrm{pH}$ was completely reversible within 20 minutes and was independent of the concentration of the buffer $(20-100 \mathrm{mM}$ ).
Partial reassociation may also be induced by elevated temperature: a wt protein solution in $50 \mathrm{mM}$ phopshate buffer or in $50 \mathrm{mM}$ BIS-TRIS propane at $\mathrm{pH} 8.0$ was heated for $1 \mathrm{~h}$ at $80^{\circ} \mathrm{C}$ and immediately injected on a Superdex column. The elution pattern indicated that $60 \%$ of the dimeric species had been converted to the octameric species.
Different behavior was observed when the same experiments were conducted with the Y41C mutant. In the GPC experiments, this mutant displayed only one peak, corresponding to the dimeric species, over the whole $\mathrm{pH}$ range examined either at 20 or at $80^{\circ} \mathrm{C}$ (filled squares in the inset of Figure $2 \mathrm{~A}$ ).
To test whether the association of wt dimers into octamers was caused by hydrophobic interactions among dimers, the polarity of the protein environment was decreased by addition of hydrophobic agents. Experiments were conducted on a protein solution brought to $\mathrm{pH} 8.0$ with $50 \mathrm{mM}$ BIS-TRIS propane buffer, to induce dissociation of octamers into dimers, followed by the addition of 5,10 and $15 \%$ methanol, ethanol and $n$-propanol and 5 and $10 \%$ of $n$-butanol; after 10 min the $\mathrm{pH}$ was brought to 4.0 with $100 \mathrm{mM}$ citrate buffer to induce oligomerization; after $20 \mathrm{~min}$ at $20^{\circ} \mathrm{C}$ the solution was injected onto the Superdex column. Neither methanol nor ethanol inhibited formation of the oligomer at $\mathrm{pH} 4.0$ : only $15 \%$ $n$-propanol or $10 \% n$-butanol was able to block formation of the oligomer.
To test whether insertion of a bulky residue at many points in the protein alters the association process induced by $\mathrm{pH}$, the protein was reacted with phenylglyossal. The reagent modified at least fourteen arginine residues per dimer out of a total of 42 : the chemically modified protein was fully active. The solution of the chemically modified protein was brought to $\mathrm{pH}$ 4.0 with citrate buffer to induce oligomerization and injected on the Superdex column: no octameric species were detected.
## Analytical ultracentrifugation and dynamic light scattering
Analytical ultracentrifugation experiments performed on the wt protein yielded two peaks, with sedimentation coefficients of $6 \mathrm{~S}$ and $18 \mathrm{~S}$, in a ratio that varied as a function of $\mathrm{pH}$ (Figure $2 \mathrm{~B}$ ). At $20^{\circ} \mathrm{C}$ and $\mathrm{pH} 6.0$, the $18 \mathrm{~S}$ species was predominant (solid line), whereas at $\mathrm{pH} 8.0$ only, the $6 \mathrm{~S}$ species was present (dashed line). The value of $18 \mathrm{~S}$ observed for the heavier species corresponds to a molecular mass of about $400 \mathrm{kDa}$, in agreement with the GPC results. The molecular mass of the lighter species is around $100 \mathrm{kDa}$, consistent with a dimeric form of the protein and also in agreement with the GPC data. The Y41C mutant exhibited only the $6 \mathrm{~S}$ peak at both $\mathrm{pH} 6.0$ and $\mathrm{pH} 8.0$ (data not shown).
The size of the recombinant amidase and of the Y41C mutant was assessed by DLS. The DLS apparatus was used to determine the hydrodynamic radius $\left(R_{\mathrm{H}}\right)$ of the photon scattering amidase molecules in buffer, by analyzing the time scale of the fluctuations in the amount of scattered light. Determination of the $R_{\mathrm{H}}$ provides an estimate of the molecular mass of the protein species under study. This estimate is useful for discriminating, for example, between a dimer and a monomer. The $\mathrm{R}_{\mathrm{H}}$ of amidase was calculated to be $4.23 \mathrm{~nm}$, according to the classical Stokes-Einstein relation for a sphere (Table 2). Software-based conversion of $\mathrm{R}_{\mathrm{H}}$ measurements estimated a molecular mass of $95.4 \mathrm{kDa}$, which accounted for an amidase dimer. This value did not change significantly, even at the lowest possible concentration attainable with the DLS instrument, ranging from 4.23 to $4.05 \mathrm{~nm}$ (molecular mass of $95.4 \mathrm{kDa}$ versus $86.3 \mathrm{kDa}$ ). A decrease in the polydispersity of the protein preparation (from 40 to $20 \%$ polydispersity) was observed by decreasing the protein concentration, but no conversion from dimer to monomer was detected, indicating that the dimeric form was the most stable one in the concentration range tested $\left(3.0-0.2 \mathrm{mg} \mathrm{ml}^{-1}\right)$. Even with a polydispersity value of $40 \%$, the measured radius perfectly fitted a monomodal gaussian distribution, not a bimodal size distribution. Therefore we can exclude the presence of species other than
Table 2. Estimation of molecular mass by dynamic light scattering measurements with the wild type (wt) amidase and the Y41C mutant. Abbreviation: $\mathrm{R}_{\mathrm{H}}=$ hydrodynamic radius.
| | $\mathrm{pH} 8.0$
$\mathrm{R}_{\mathrm{H}}(\mathrm{nm})^{1}$ | $\mathrm{pH} 4.0$
$\mathrm{R}_{\mathrm{H}}(\mathrm{nm})^{1}$ | $\mathrm{pH} 8.0$
Estimated molecular mass
$(\mathrm{kDa})^{2}$ | $\mathrm{pH} 4.0$
Estimated molecular mass
$(\mathrm{kDa})^{2}$ |
| :---: | :---: | :---: | :---: | :---: |
| Wt amidase | 4.2 | 7.5 | 95.4 | 385.7 |
| Y41C mutant | 4.4 | 4.5 | 106.0 | 111.0 |
${ }^{1}$ Mean hydrodynamic radius derived from the measured translational diffusion coefficient using the Stokes-Einstein equation.
${ }^{2}$ Molecular mass estimated from $\mathrm{R}_{\mathrm{H}}$ assuming that the particles are spherical and of standard density.
the dimer. At $\mathrm{pH} 4.0, \mathrm{R}_{\mathrm{H}}$ increased to $7.52 \mathrm{~nm}$ (with a polydispersity of less than $20 \%$ ), indicating an oligomeric state of amidase which contained more than one dimer. This value corresponded to an estimated molecular mass of $385.7 \mathrm{kDa}$, compatible with a tetramer of dimers (Table 2). In contrast, light scattering experiments with the Y41C mutant, conducted at a protein concentration ranging from 3.0 to $0.2 \mathrm{mg} \mathrm{ml}^{-1}$ either at $\mathrm{pH} 4.0$ or 8.0 , always yielded the presence of a species compatible with a dimer (Table 2). Also in the DLS experiments, the measured radii fitted a monomodal gaussian distribution, not a bimodal size distribution.
## Monomer detection
To determine if the dimers could be chemically dissociated, solutions of the wt and mutant proteins in $50 \mathrm{mM}$ BIS-TRIS propane buffer $\mathrm{pH} 8.0$ (having only dimers) were treated with urea (up to $2 \mathrm{M}$ ), guanidinium chloride (up to $0.8 \mathrm{M}$ ) or thiocyanate (up to $0.5 \mathrm{M}$ ) (Ptitsyn et al. 1990). At all concentrations the protein maintained full activity and when injected on the Superdex column, no dissociation of the dimer occurred. Likewise, incubation of the two proteins in $50 \mathrm{mM}$ Tris adjusted to $\mathrm{pH} 11.0$ (Almatawah et al. 1999) did not cause dissociation of the dimers.
## Spectroscopic analyses
To assess whether the behavior of the wt and Y41C proteins as a function of $\mathrm{pH}$ was accompanied by $\mathrm{pH}$-dependent conformational changes, circular dichroism (CD) and intrinsic fluorescence emission spectra of both proteins were recorded at $\mathrm{pH}$ 5.0 and 8.0 (Figure 3). A significant modification of the tertiary and secondary structure of the wt protein occurred on transition between these $\mathrm{pH}$ values (Figures $3 \mathrm{~A}$ and $\mathrm{B}$ ) as indicated by the decrease in fluorescence (Figure 3A) and ellipticity (Figure 3B). No structural modifications in the Y41C mutant were detected (Figures 3C and D).
The experiments with ANS reported in Figure 4, show that the extrinsic fluorescence emission spectrum of wt protein with ANS at $\mathrm{pH} 5.0$ was almost twice that measured at $\mathrm{pH} 8.0$, whereas the spectrum of the Y41C mutant was only slightly increased (Figures 4A and B). These results demonstrate that the dye bound more tightly to the wt protein at $\mathrm{pH} 5.0$ than at $\mathrm{pH} 8.0$, whereas $\mathrm{pH}$ had no effect on the binding of dye to the Y41C mutant protein.
## Thermoactivity, thermostability and thermal unfolding
Thermoactivity and thermostability of the wt and Y41C proteins were studied at $\mathrm{pH} 5.0$ and 8.0. Thermoactivity of the two proteins at both $\mathrm{pH} 5.0$ and $\mathrm{pH} 8.0$ was similar, with a maximum at $95^{\circ} \mathrm{C}$ (data not shown). Thermal stability of the wt enzyme and its mutant $\mathrm{Y} 41 \mathrm{C}$ at $\mathrm{pH} 5.0$ and $70^{\circ} \mathrm{C}$, were similar, with half-life values over $70 \mathrm{~h}$ (Table 3). At $80^{\circ} \mathrm{C}$ no differences were detected when both proteins were tested at $\mathrm{pH} 5.0$, but at $\mathrm{pH} 8.0$, the half-life of the wt protein was reduced by more than half, whereas that of the mutant was reduced by more than $99 \%$.
The DSC profile of both proteins is shown in Figure 5. Thermal unfolding of the wt protein at $\mathrm{pH} 8.0$ gave a $T_{\mathrm{m}}$ of $94.4^{\circ} \mathrm{C}$. At $\mathrm{pH} 5.0$ the wt protein showed a $T_{\mathrm{m}}$ of $104.3^{\circ} \mathrm{C}$. The Y41C mutant showed lower $T_{\mathrm{m}}$ values at both $\mathrm{pH} 8.0\left(83.5^{\circ} \mathrm{C}\right)$ and $\mathrm{pH} 5.0\left(97.3^{\circ} \mathrm{C}\right)$. No reversibility was found under the measured conditions.
## Modeling of three dimensional structure
The 3D structure of SSAM was obtained by homology modeling. The selected template that best aligns with the target sequence is PAM from Stenotrophomonas maltophila (Labahn et al. 2002). Figure 6 shows the sequence alignment of SSAM, MAE2, FAAH and PAM, focusing on the highly conserved signature sequence and the active site (shown in color). The Ser-Ser-Lys catalytic triad is highlighted in red. After modeling the target on the template, the 3D structure of SSAM results in an alpha/beta protein with a Root Mean Square Deviation to PAM equal to $0.08 \mathrm{~nm}$. The content of secondary structure is characterized by $25.7 \%$ of alpha, $12.2 \%$ of beta and $62.1 \%$ of coil structural types, respectively, in good agreement with the percentage calculated from the far UV CD spectra.
The putative interaction patches localized on the accessible surface of the protein were predicted with ISPRED, a neuralnetwork based predictor for protein interaction sites that has been described previously (Fariselli et al. 2002). This allowed us to compute the protein putative dimer with ZDOCK (Chen and Weng 2002), a program based on complementarity shape and electrostatic interactions. Figure 7 shows the putative model of the SSAM dimer: charged ion-pair residues putatively involved in the electrostatic interactions between the two monomers are highlighted as red and pink spheres (negatively charged) and blue and magenta spheres (positively
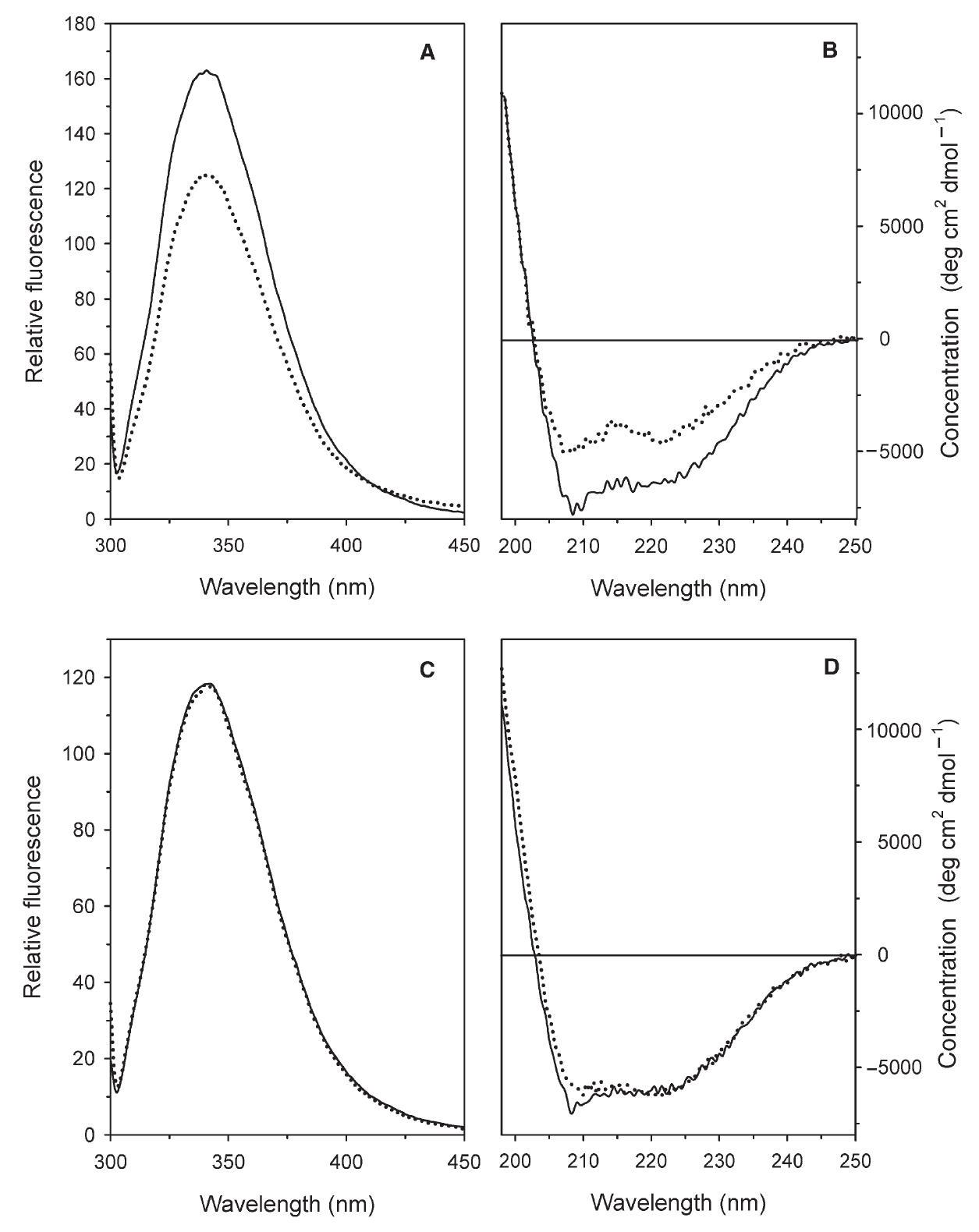
Figure 3. Effects of $\mathrm{pH}$ on the intrinsic fluorescence and on far UV CD spectra of the Solfolobus solfataricus recombinant wild type (wt) amidase and the Y41C mutant. Fluorescence emission spectra (A, C) were recorded with the excitation wavelength at $295 \mathrm{~nm}$. Far UV CD spectra were recorded in a $0.1 \mathrm{~cm}$ path-length quartz cuvette. All spectra were recorded with $0.1 \mathrm{mg} \mathrm{ml}^{-1}$ protein concentrations at $20^{\circ} \mathrm{C}$ after overnight incubation of the wt $(\mathrm{A}, \mathrm{B})$ and the $\mathrm{Y} 41 \mathrm{C}$ mutant $(\mathrm{C}, \mathrm{D})$ at $\mathrm{pH} 8.0$ ( $20 \mathrm{mM}$ sodium phosphate, solid line) and $\mathrm{pH} 5.0(20 \mathrm{mM}$ sodium citrate, dotted line).
charged). The model also shows the catalytic triad (K96 (orange), S171 (yellow) and S195 (green)) at the active site, E337 (light blue) and D442 (red) at the entrance of the active site and Y41 in brilliant green. It is evident that, according to the model, Y41 is not involved in the interaction surface between monomers in the dimer. This is in agreement with the finding that the Y41C mutation does not affect protein dimerization but only the oligomerization of dimers.
## Discussion
The most interesting feature of the recombinant wt amidase was its response to $\mathrm{pH}$. The octameric species was predominant at acidic $\mathrm{pH}$ values, whereas the dimeric species was predominant at $\mathrm{pH}$ values higher than 7.0. This behavior is infrequent, with most proteins behaving in the opposite way, i.e., the association of protomers in multimers is impaired at acidic pH values (Ralston 1991, Zeng et al. 1997, Calvete et al. 1999, Hochgrebe et al. 2000, Madern et al. 2000, Madern et al. 2001, Wah et al. 2001, Lew et al. 2003).
Our results suggest that the association of dimers into octamers is the consequence of conformational changes induced by the protonation of some ionizable group allowing the interaction of the segment containing Y41 (LLKLQLESYERLDSLP) with a hydrophobic patch exposed to solvent in the contiguous dimer as proposed in the scheme reported in Figure 8 . Reducing the polarity of the environment by organic solvents such as $n$-butanol or $n$-propanol decreased the hydrophobic interactions and increased the inhibition of the dimer association in accordance with the effects of organic solvents on most enzymes (Williams et al. 1995).
The experimental results lead to the conclusion that acidic
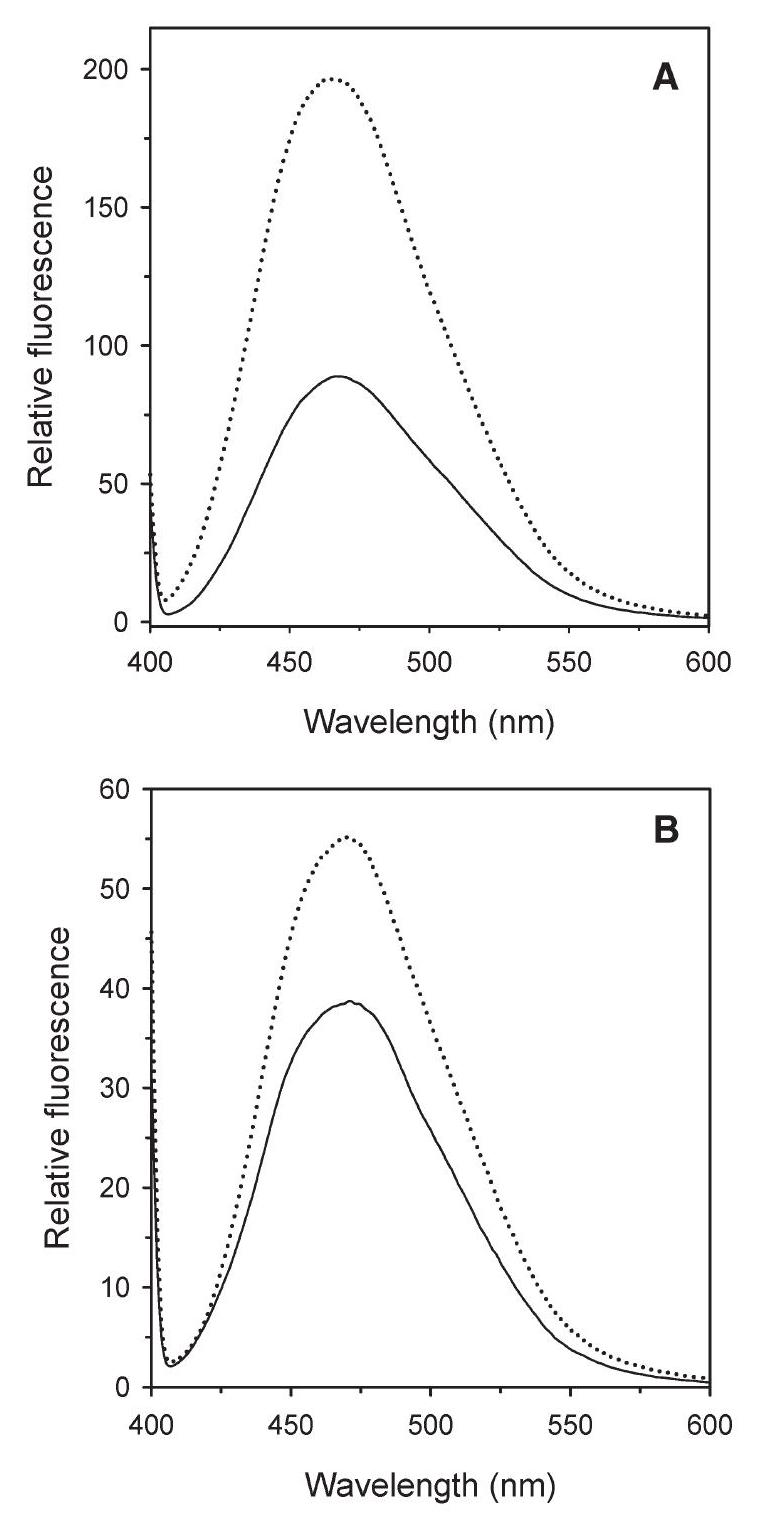
Figure 4. Relative accessibility of hydrophobic residues in Solfolobus solfataricus wild type (wt) amidase (A) and its Y41C mutant (B) monitored by the extrinsic fluorescence of 8 -anilinonaphtalene-1-sulfonic acid ammonium salt (ANS). The ANS $(25 \mu \mathrm{M})$ was added to the protein $(2.5 \mu \mathrm{M})$, which had previously been incubated overnight at $\mathrm{pH} 8.0$ (20 mM sodium phosphate, solid line) and at $\mathrm{pH} 5.0(20 \mathrm{mM}$ sodium citrate, dotted line). Fluorescence emission spectra (390 nm) were recorded at $20^{\circ} \mathrm{C}, 10 \mathrm{~min}$ after the addition of ANS.
Table 3. Thermal stability (half-lives, h) of the wild type (wt) Solfolobus solfataricus amidase and its Y41C mutant measured at $\mathrm{pH} 5.0$ and 8.0.
| $\mathrm{pH}$ | | $70^{\circ} \mathrm{C}$ | $80^{\circ} \mathrm{C}$ | $90^{\circ} \mathrm{C}$ |
| :--- | :--- | :---: | :---: | :---: |
| 5.0 | $\mathrm{WT}$ | $>70$ | 65 | 60 |
| | $\mathrm{Y} 41 \mathrm{C}$ | $>70$ | 64 | 20 |
| 8.0 | $\mathrm{WT}$ | $>70$ | 25 | 0.5 |
| | $\mathrm{Y} 41 \mathrm{C}$ | $>70$ | 0.5 | 0.25 |
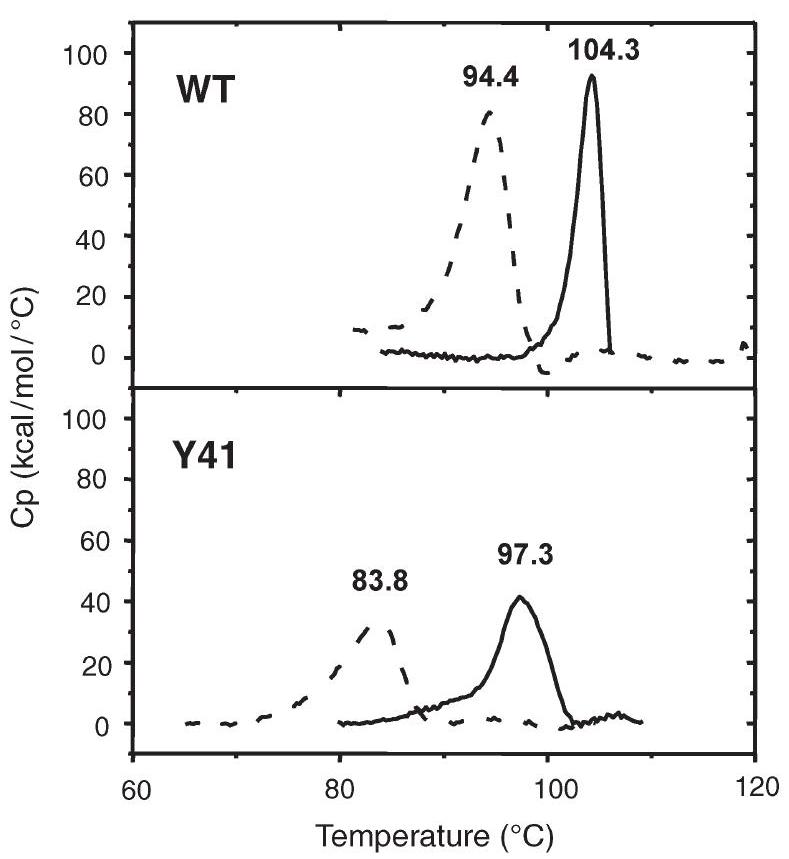
Figure 5. Differential scanning calorimetry thermograms of wild type (wt) amidase and its Y41C mutant at different $\mathrm{pH}$ values. Solid lines correspond to traces obtained in $20 \mathrm{mM}$ citrate buffer at $\mathrm{pH} 4.0$, dotted lines to those obtained in $20 \mathrm{mM}$ sodium phosphate at $\mathrm{pH}$ 8.0. The concentration was always expressed as moles of dimer. Denaturation temperatures $\left(T_{\mathrm{m}}\right)$ are indicated above the corresponding peak.
$\mathrm{pH}$ may induce changes in the tertiary structure of the wt protein (as revealed by fluorescence and CD spectra) which acquires a new spatial arrangement that permits new hydrophobic interactions. The conformational changes induced in the dimers may lead to the exposure on their surface of sticky hydrophobic segments that could result in hydrophobic zipper-like structures, as reported for proteins such as ERK2 (Khokhlatchev et al. 1997), PAK-1 (Hoffman and Cerione 2000) and sulfotransferases (Petrotchenko et al. 2001), and in the $\mathrm{pH}$-dependent transition of viral fusion proteins (Hsu et al. 2002, Yao et al. 2003). On this basis, the interactions among the dimers would then be mainly hydrophobic and Y41 could have the role of a trigger priming oligomerization. In turn, oligomerization would be impaired when Y41 is substituted with a cysteine residue, as in the Y41C mutant.
Such an interpretation is supported by the fluorescence experiments with ANS and by the chemical inhibition of oligomerization. The fluorescence experiments with ANS demonstrate a higher availability of hydrophobic residues in the wt protein at acidic $\mathrm{pH}$ values. Because ANS binds to hydrophobic groups of a protein (Anderson and Weber 1966), in the wt protein, at $\mathrm{pH} 5.0$, more hydrophobic groups should be available for interaction with the dye, i.e., those groups embedded in the core of the protein at $\mathrm{pH} 8.0$ are more exposed at $\mathrm{pH} 5.0$.
The association of wt amidase dimers into octamers, as induced by acidic $\mathrm{pH}$ was hampered by reduced environmental polarity and by the insertion of a bulky residue such as phenylglyossal in many points of the protein, which could modify the
| SSAM | GKLKGKRICI KDNVMIAGIPMLN GSKMLEGFVPHMDATVVSRILDEAGEIVAK TTCEDLCFSGGSHTSYPWP |
| :---: | :---: |
| MAE2 | GPLRGIAVGI KDIIDTANMPTEM GSEIYRGWQPRS DAPVVMMLKRAGATIIGK TTTTAFASRDPTATLN--- |
| FAAH | DSTL GLSLNEGVPAEC DSVVVHVLKLQGAVPFVH TNVPQSMFSYDCSNPLFG- |
| PAM | GPLHGIPLLLKDNINAAPMATSA GSLALQGFRP-DDAYLVRRLRDAGGAVVLGK TNLSEWANFRGNDSISGWS |

Figure 6. Sequence alignment of the conserved stretch. The 130 amino acids, including the "amidase signature sequence" of Sulfolobus solfataricus amidase (SSAM) (GB AF290611), malonyl amidase E2 (MAE2) from Bradyrhizobium japonicum (PDB 1GR8), fatty acid amide hydrolase (FAAH) from Rattus norvegicus (PDB 1MT5) and peptidyl amidase (PAM) from Stenotrophomonas maltophila (PDB 1M22). Invari ant residues are reported in green whereas conservative residues are in blue. The catalytic triad $(\mathrm{K}, \mathrm{S}, \mathrm{S})$ is indicated in red.
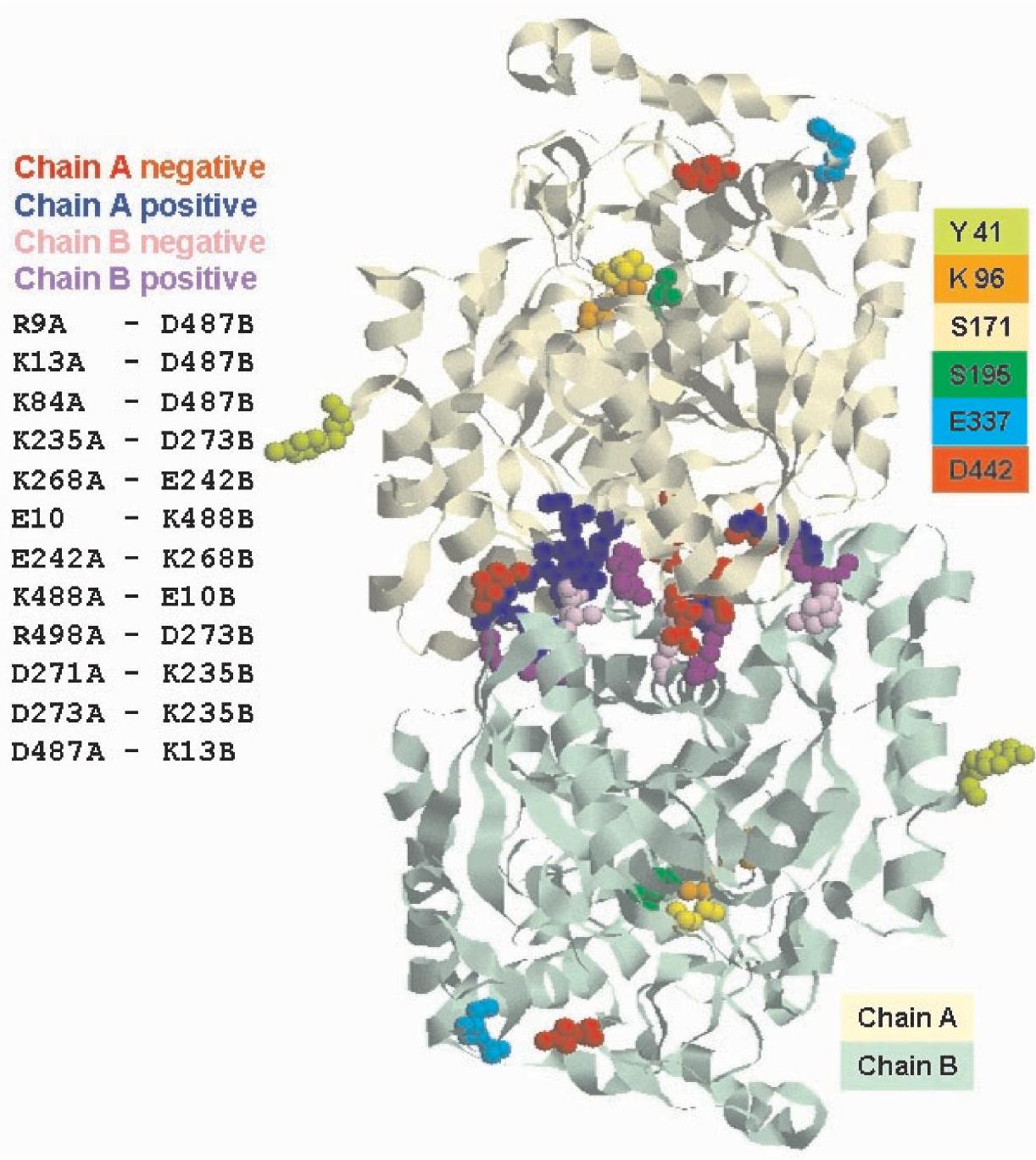
Figure 7. Model of the Sulfolobus solfataricus amidase (SSAM) dimer. Putative model of the SSAM dimer: charged ion-pair residues putatively involved in the electrostatic interactions (positively charged groups are blue and magenta, negatively charged groups are red and pink) between the two monomers are highlighted. Also shown are the catalytic triad (K96 (orange), S171 (yellow) and S195 (green)) at the active site, E337 (light blue) and D442 (red) at the entrance of the active site and Y41 in brilliant green.
conformation of dimers, rendering them unable to associate into octamers without modifying the specific activity of the enzyme.
The molecular mass of the SSAM as determined by the amino acid sequence is $55,784 \mathrm{Da}$. Ultracentrifugation analyses yielded two peaks with $\mathrm{S}_{\mathrm{w}, 20}=6 \mathrm{~S}$ and $18 \mathrm{~S}$ corresponding to a molecular mass of about $100 \mathrm{kDa}$ and $400 \mathrm{kDa}$, respectively, which are compatible with a dimeric and an octameric species. These values were confirmed by the DLS and GPC experiments.
Attempts failed to dissociate the dimer of the wt protein and the Y41C mutant with urea or guanidinium chloride (generic

Figure 8. Proposed scheme of the Sulfolobus solfataricus amidase (SSAM) octamer. Each sphere represents a dimer. Hydrophobic residues are in magenta. Also shown are E337 (red) and D442 (light blue) at the entrance to the active site and $\mathrm{Y} 41$ in green.
chaothropes that affect the hydrogen bond network of a protein), a lipophilic salt such as thiocyanate (which can interfere with ion pairs located either on the surface or in the hydrophobic core of folded proteins (Ptitsyn et al. 1990)) or with treatment at $\mathrm{pH} 11.0$ (Almatawah et al. 1999), suggesting that the two subunits are strongly bound, likely through the electrostatic interactions depicted in the dimer model of Figure 7.
Maximal activity of the recombinant wt amidase and its mutant $\mathrm{Y} 41 \mathrm{C}$ was found at $\mathrm{pH}$ 5.0. Aromatic substrates are better than aliphatic substrates for both enzymes, as reflected by the Michaelis constant $\left(K_{\mathrm{m}}\right)$ values, for which aliphatic substrates were one order of magnitude higher than aromatic substrates. The aliphatic substrates entered the active site at $\mathrm{pH} 5.0$ with greater difficulty, but the products left it more easily, as reflected by the higher turnover rate $\left(k_{\text {cat }}\right)$ relative to aromatic substrates. However, $k_{\text {cat }}$ values for aliphatic substrates are higher than those for aromatic substrates. As a consequence, enzymatic efficiency (expressed as $k_{\text {cat }} / K_{\mathrm{m}}$ ) of both proteins was high both with aliphatic and aromatic substrates, but was higher with the latter. These results, taken together with the modeling data reported in Figure 7 and 9, suggest that at the active site of the enzyme, hydrophobic residues achieve a better interaction with aromatic substrates and some carboxylic groups which, with their negative charges, may drive away the negatively charged product of the reaction.
As expected for proteins from a hyperthermophilic organism, thermal stability for both proteins, reported in Table 3 as half-lives and in Figure 5 as $T_{\mathrm{m}}$ values, was high (a decrease in activity was observed only in the mutant at $90^{\circ} \mathrm{C}$, as expected in a protein with an additional cysteine) but differed with $\mathrm{pH}$. As shown by GPC, DLS and AUC, the octameric species is predominant in the wt protein at acidic $\mathrm{pH}$ values, whereas the dimeric species is predominant at $\mathrm{pH}$ values higher than 7.0. In Y41C, only the dimeric species was present at all $\mathrm{pH}$ values.
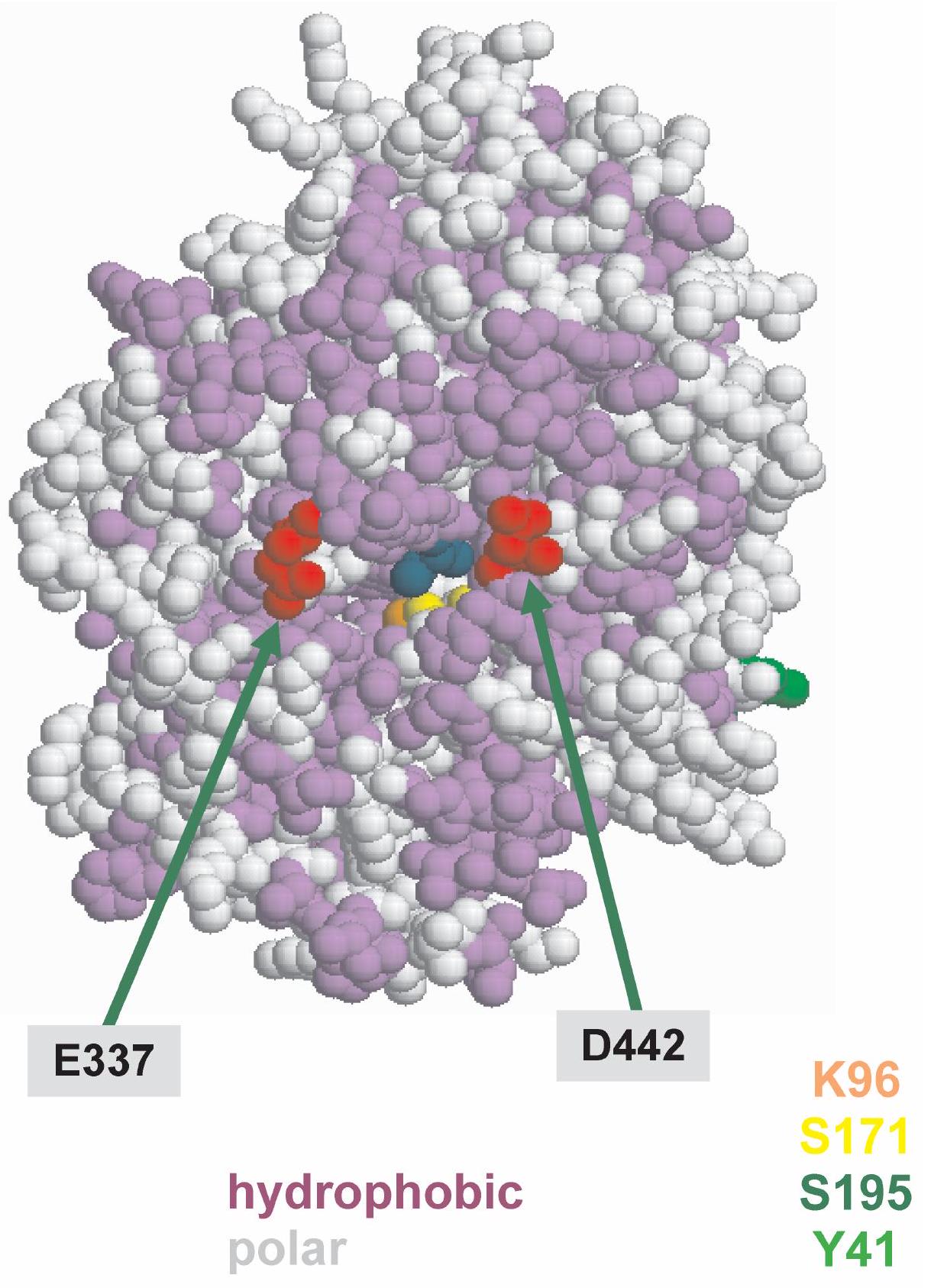
Figure 9. Hydrophobic and charged residues around the entrance of the Sulfolobus solfataricus amidase active site.
Combining all the information acquired through spectroscopic analyses, GPC, AUC, DLS and DSC experiments, we draw the following conclusions.
The Y41C dimer is less compact than the wt dimer. The thermal stability of $\mathrm{Y} 41 \mathrm{C}$ at $80^{\circ} \mathrm{C}$ and $\mathrm{pH} 8.0$ is reduced to a half-life of $0.5 \mathrm{~h}$ compared with $25 \mathrm{~h}$ for the wt at the same $\mathrm{pH}$ and temperature. However, when Y41C is brought to $\mathrm{pH} 5.0$, it assumes a structure and demonstrates behavior comparable to that of the wt protein at the same $\mathrm{pH}$ and temperature with a thermal stability of $64 \mathrm{~h}$, although no octamers are formed. Microcalorimetry indicated that, at $\mathrm{pH} 4.0, \mathrm{Y} 41 \mathrm{C}$ has a $T_{\mathrm{m}}$ of $97.3^{\circ} \mathrm{C}$, which is close to that of the dimeric wt species at $\mathrm{pH}$ $8.0\left(94.4^{\circ} \mathrm{C}\right)$. At $\mathrm{pH} 4.0$ to $5.0, \mathrm{Y} 41 \mathrm{C}$, but not the wt protein, becomes more compact, resulting in increased ANS binding, although this was not detected spectrophotometrically.
In contrast, in the wt protein, acidic $\mathrm{pH}$ (4.0 or 5.0), as shown by GPC, AUC and DLS, lead to oligomerization, accompanied by the highest $T_{\mathrm{m}}\left(104.4^{\circ} \mathrm{C}\right)$, changes in tertiary structure, as revealed by fluorescence and CD spectra, and larger amounts of ANS binding the protein.
At $\mathrm{pH} 5.0$ or $\mathrm{pH} 8.0$ both proteins were active up to $95^{\circ} \mathrm{C}$. This means that the dimer of $\mathrm{Y} 41 \mathrm{C}$ (present at all $\mathrm{pH}$ values), the dimer of the wt (present at $\mathrm{pH} 8.0$ ) and the octamer of the wt predominant at $\mathrm{pH} 5.0$, are all thermoactive species.
We speculate that oligomerization could have a physiological role in the cell which exists in a highly acidic environment
$\left(\mathrm{pH} 2.5\right.$ ) and at an average temperature of about $85^{\circ} \mathrm{C}$. The inner $\mathrm{pH}$ of the cell is 6.8 (De Rosa et al. 1975, Moll and Shafer 1988) but this value may not be constant because most of the cell's energy is employed in contending with the acidic environment. Temperature too, could fluctuate owing to the discontinuous emission of hot gases. Therefore, oligomerization through an increase in hydrophobic interactions induced by $\mathrm{pH}$ and temperature could be one of the mechanisms increasing protein thermostability. Other examples of increased protein thermostability through oligomerization were suggested by Opitz et al.(1987) on the basis of fragmentation studies on lactate dehydrogenase, from modeling studies by Kohlhoff et al. (1996) on triosephosphate isomerase, by Villeret et al. (1998) on ornithine carbamoyltransferase and more recently by Shima et al. (2000) on formyltransferase from the hyperthermophilic Methanopyrus kandleri.
## Acknowledgments
We are grateful to Prof. E. Chiancone for her criticism and helpful discussion and to Prof. B. Maras for amino acid determinations. This research was funded by Italian MIUR (Ministery of University and Research), by CNR Target Project on Biotechnology and by ASI (Italian Space Agency).
## References
Almatawah, A.A., R. Cramp and A. Cowan. 1999. Characterization of an inducible nitrilase from a thermophilic bacillus. Extremophiles 3:283-291.
Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D.J. Lipman. 1997. Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
Anderson, S. and G. Weber. 1966. The reversibile acid dissociation and hybridization of lactic dehydrogenase. Arch. Biochem. Biophys. 116:207-223.
Bracey, M.H., M.A. Hanson, K.R. Masuda, R.C. Stevens and B.F. Cravatt. 2002. Structural adaptation in a membrane enzyme that terminates endocannabinoid signaling. Science 296: $1793-1796$.
Calvete, J.J., H.H. Thole, M. Raida, C. Urbane, A. Romero and T.B. Grangeiro. 1999. Molecular characterization and crystallization of Diocleinae lectins. Biochim. Biophys. Acta 1430:367-375.
Chebrou, H., F. Bigey, A. Arnaud and P. Galzy. 1996. Study of the amidase signature group. Biochim. Biophys. Acta 1298:285-293.
Chen, R. and Z. Weng. 2002. Docking unbound proteins using shape complementarity, desolvation and electrostatics. Proteins 47: 281-294
Cravatt, B.F., D.K. Giang, S.P. Mayfield, D.L. Boger, R.A. Lerner and N.B. Gilula. 1996. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83-87.
Curnow, A.W., K. Hong, R. Yuan, S. Kim, O. Martins, W. Winkler, T.M. Henkin and D. Soll. 1997. Glu-tRNA ${ }^{\text {Gln }}$ amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc. Natl. Acad. Sci. USA. 94: $11819-11826$.
De Rosa, M., A. Gambacorta and D. Bulock. 1975. Extremely thermophilic acidophilic bacteria convergent with Sulfolobus acidocaldarius. J. Gen. Microbiol. 86:154-164.
Fariselli, P., F. Pazos, A. Valencia and R. Casadio. 2002. Prediction of protein-protein interaction sites in heterocomplexes with neural networks. Eur. J. Biochem. 269:1356-1361.
Gaffney, T.D., O. da Costa e Silva, T. Yamada and T. Kosuge. 1990. Indolacetic acid operon of Pseudomonas syringae subsp. savastanoi: transcription analysis and promoter identification. J. Bacteriol. 172:5593-5601.
Gill, S. and P.H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182: $319-326$.
Gomi, K., K. Kitamoto and C. Kumagai. 1991. Cloning and molecular characterization of the acetamidase-encoding gene (amdS) from Aspergillus orizae. Gene 108:91-98.
Hochgrebe, T., G.J. Pankhurst, J. Wilce and S.B. Easterbrook-Smith. 2000. $\mathrm{pH}$-dependent changes in the in vitro ligand-binding properties and structure of human clusterin. Biochemistry 15:1411-1419.
Hoffman, G.R. and R.A. Cerione. 2000. Flipping the switch: the structural basis for signaling through the CRIB motif. Cell 102: 403-406.
Hsu, C.H., S.H. Wu, D.K. Chang and C. Chen. 2002. Structural characterizations of fusion peptide analog of influenza virus hemagglutinin. Implication of the necessity of a helix-hinge-helix motif in fusion activity. J. Biol. Chem. 277:22,725-22,733.
Khokhlatchev, A., S. Xu, J. English, P. Wu, E. Schaefer and M.H. Cobb. 1997. Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J. Biol. Chem. 272:11,057-11,062.
Kohlhoff, M., A. Dahm and R. Hensel. 1996. Tetrameric triosephosphate isomerase from hyperthermophilic Archaea. FEBS Lett. 383:245-250
Koutek, B., G.D. Prestwich, A.C. Howlett, S.A. Chin, D. Salehani, N. Akhavan and D.G. Deutsch. 1994. Inhibitors of arachidonoyl ethanolamide hydrolysis. J. Biol. Chem. 269:22,937-22,940.
Labahn, J., S. Neumann, G. Buldt, M.R. Kula and J. Granzin. 2002. An alternative mechanism for amidase signature enzymes. J. Mol. Biol. 322:1053-1064.
Leatherbarrow, R.J. 1990. Use of nonlinear regression to analyze enzyme kinetic data: application to situations of substrate contamination and background subtraction. Anal. Biochem. 184:274-278.
Lew, S., G. A. Caputo and E. London. 2003. The effect of interactions involving ionizable residues flanking membrane-inserted hydrophobic helices upon helix-helix interactions. Biochemistry 42 : 10833-10842.
Madern, D., C. Ebel, M. Mevarech, S.B. Richard, C. Pfister and G. Zaccai. 2000. Insights into the molecular relationship between malate and lactate dehydrogenases:structural and biochemical properties of monomeric and dimeric intermediates of a mutant of tetrameric L-(LDH-like) malate dehydrogenase from the halophilic archaeon Haloarcula marismortui. Biochemistry 39: $1001-1010$.
Madern, D., C. Ebel, H.A. Dale, T. Lien, I.H. Steen, N.K. Birkeland and G. Zaccai. 2001. Differences in the oligomeric states of the LDH-like MalDH from the hyperthermophilic archaea Methanococcus jannaschii and Archaeoglobus fulgidus. Biochemistry 40: $10310-10316$.
Mayaux, J.F., E. Cerebelaud, F. Soubrier, P. Yeh, F. Blanche and D. Petre. 1991. Purification, cloning, and primary structure of new enantiomer-selective amidase from Rhodococcus strain: structural evidence for a conserved genetic coupling with nitrile hydratase. J. Bacteriol. 173:6694-6704.
Moll, R. and G. Shafer, 1988. Chemiosmotic $\mathrm{H}^{+}$cycling across the plasma membrane of the thermoacidophilic archaebacterium Sulfolobus acidocaldarius. FEBS Lett. 32:359-363.
Opitz, U., R. Rudolph, R. Jaenicke, L. Ericsson and H. Neurath. 1987. Proteolytic dimers of porcine muscle lactate dehydrogenase characterization, folding and reconstitution of the truncated and nicked polypeptide chain. Biochemistry 26:1399-1406
Patricelli, M.P. and B.F. Cravatt. 2000. Clarifying the catalytic roles of conserved residues in the amidase signature family. J. Biol. Chem. 275:19177-19184.
Petrotchenko, E.V., L.C. Pedersen, C.H. Borchers, K.B. Tome and M. Negishi. 2001. The dimerization motif of cytosolic sulfotransferases. FEBS Lett. 490:39-43.
Ptitsyn, O.B., R.H Pain, G.V. Semisotnov, E. Zerovnik and O.I. Razguliaiev. 1990. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 262:20-24.
Ralston, G.B. 1991. Temperature and $\mathrm{pH}$ dependence of the self-association of human spectrin. Biochemistry 30:4179-4186.
Riordan, J. and R. Scandurra. 1972. Essential arginyl residues in aspartate aminotransferases. Bioch. Biophys. Res. Comm. 66: $417-424$.
Sali, A. and T. L. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779-815.
Schon, A., C.G. Kannangara, S. Gough and D. Soll. 1998. Protein biosynthesis in organelles requires misaminoacylation of tRNA. Nature 331:187-190.
Schuck, P. and P. Rossmanith. 2000. Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 54:328-341
Scotto d'Abusco, A., S. Ammendola, R. Scandurra and L. Politi. 2001. Molecular and biochemical characterization of the recombinant amidase from hyperthermophilic archaeon Sulfolobus solfataricus. Extremophiles 5:183-192.
Shima, S., R.K. Thauer, U. Ermler, H. Durchschlag, C. Tziatzios and D. Shubert. 2000. A mutation affecting the association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri and its influence on the enzyme's activity and thermostability. Eur. J. Biochem. 267:6619-6623.
Shin, S., Y.S. Yun, H.M. Koo, Y.S. Kim, K.Y. Choi and B.H. Oh. 2003. Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad. J. Biol. Chem 278:24937-24943.
Villeret, V., B. Clantin, C. Tricot, C. Legrain, M. Roovers, V. Stalon, N. Glansdorff and J. Van Beeumen. 1998. The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures. Proc. Natl. Acad. Sci. USA 95:2801-2806.
Wah, D.A., A. Romero, F. Gallego del Sol, B.S. Cavad, M.V. Ramos, T.B. Grangeirp, A.H. Sampaio and J.J. Calvete. 2001. Crystal structure of native an $\mathrm{Cd} / \mathrm{Cd}$-substituted Dioclea guaianensis seed lectin. A novel manganese-binding site and structural basis of dimer-tetramer association. J. Mol. Biol. 310:885-894.
Williams, D.L Jr., I. Rapanovich and A. Russel. 1995. Proteins in essential nonaqueous environments. In Protein-Solvent Interactions. Ed. R.B. Gregory. Marcel Dekker, New York, pp 327-341.
Yao, Y., K. Ghosh, R.F. Epand, R.M. Epand and H.P. Ghosh. 2003 Membrane fusion activity of vesicular stomatitis virus glycoprotein $\mathrm{G}$ is induced by low $\mathrm{pH}$ but not by heat or denaturant. Virology $310: 319-332$.
Zeng, X., H. Zhu, H.A. Lashuel, J.C. Hu. 1997. Oligomerization properties of GCN4 leucine zipper e and $\mathrm{g}$ position mutants. Protein Sci. 6:2218-2226.

Journal of
Signal Transduction

International Journal of Microbiology
